




The mechanisms driving abnormal pro-inflammatory responses to cigarette smoke in COPD remain unclear. MicroRNA (miR)-320d was previously shown to have anti-inflammatory effects, being upregulated by inhaled corticosteroids. Therefore, our objective was to study whether miR-320d suppresses smoke-induced airway epithelial pro-inflammatory responses and if this is compromised in COPD.
MethodsWe investigated the anti-inflammatory mechanisms of miR-320d in cigarette smoke extract (CSE)-exposed primary bronchial epithelial cells (PBECs), comparing COPD and control cells using a miR-320d mimic. Additionally, we assessed whether miR-320d expression is altered with COPD and its severity, investigating lung tissue from two independent cohorts of non-COPD controls (non/current/ex-smokers) and COPD patients (current/ex-smokers) with mild/moderate and severe disease.
ResultsMiR-320d overexpression attenuated baseline and CSE-induced pro-inflammatory CXCL8, IL-1α and GM-CSF secretion in non-COPD-derived PBECs. This effect was not observed for CXCL8 and IL-1α in COPD-derived PBECs. RNA-sequencing showed that miR-320d significantly regulates the expression of 137 genes in CSE-exposed epithelium, the upregulated genes being enriched in “interleukin-33-mediated signaling” and the downregulated genes in “response to cytokine” (including IRAK1) pathways. Higher miR-320d levels were associated with lower IRAK1 expression in control but not COPD-derived PBECs. Finally, miR-320d levels were lower in lung tissue of COPD patients vs non-smoking controls and in severe vs mild/moderate COPD patients.
ConclusionsmiR-320d's suppressive effect on bronchial epithelial pro-inflammatory responses cells may be compromised in COPD. Additionally, miR-320d expression in lung tissue was lower with COPD severity. Thus, lower miR-320d anti-inflammatory action may contribute to persisting inflammation in COPD.

Chronic obstructive pulmonary disease (COPD) is characterized by chronic airway inflammation, airway remodeling and/or destruction of the alveoli (emphysema), leading to airflow limitation [1]. The inhalation of noxious particles and gases, including cigarette smoke, and genetic susceptibility are major risk factors for the development of COPD [2]. The symptoms of COPD can be reduced by the use of inhaled corticosteroids (ICS), but not all patients are responsive to ICS. More insight into underlying mechanisms is needed in order to identify novel treatments strategies. The airway epithelium forms the first barrier to the inhaled environment, including noxious particles and gases, and plays a crucial role in host defense and maintaining lung homeostasis [3]. Exposure to cigarette smoke damages the airway epithelium, leading to the secretion of several cytokines and chemokines, including C-X-C Motif Chemokine Ligand 8 (CXCL8), interleukin (IL)-1α and granulocyte macrophage-colony stimulating factor (GM-CSF) [4]. CXCL8 attracts neutrophils, IL-1α activates pro-inflammatory pathways including NF-κB signaling and GM-CSF regulates the differentiation, activation and survival of neutrophils, eosinophils and monocytes/macrophages [4–6]. Higher levels of CXCL8, IL-1α and GM-CSF have been observed in sputum of COPD patients compared to (non-)smoking controls [5–7]. However, the molecular mechanisms that drive these abnormal pro-inflammatory responses in airway epithelium of COPD patients in response to cigarette smoke remain unclear.
Previous studies have shown that microRNAs (miRNAs), 19–21 nucleotide-short noncoding RNA molecules that regulate gene expression, can contribute to the pathogenesis of COPD [8–10]. In lung tissue and sputum of COPD patients altered miRNA expression profiles have been found compared to healthy controls [11–13]. Furthermore, microRNA-320d (miR-320d) expression was found to be increased in bronchial biopsies of COPD patients after using ICS [14]. In vitro, ICS treatment upregulated miR-320d expression in air-liquid interface-differentiated primary bronchial epithelial cells (PBECs) from healthy controls. Furthermore, transfection with miR-320d mimic reduced CXCL8 levels upon cigarette smoke extract (CSE) exposure in normal airway epithelium [14].
In this study, we hypothesized that (1) miR-320d suppresses cigarette smoke-induced pro-inflammatory responses in the airway epithelium and (2) that this suppression is compromised in COPD. To assess anti-inflammatory effects of miR-320d and compare between COPD patients and control, PBECs were transfected with mimic miR-320d and effects on pro-inflammatory cytokine release and transcriptional profiles were investigated upon CSE stimulation. To determine whether miR-320d is differentially expressed with COPD in peripheral lung tissue, we compared two independent cohorts of never smokers as well as never/current/former smokers without airflow limitation or with GOLD stage II-IV COPD.
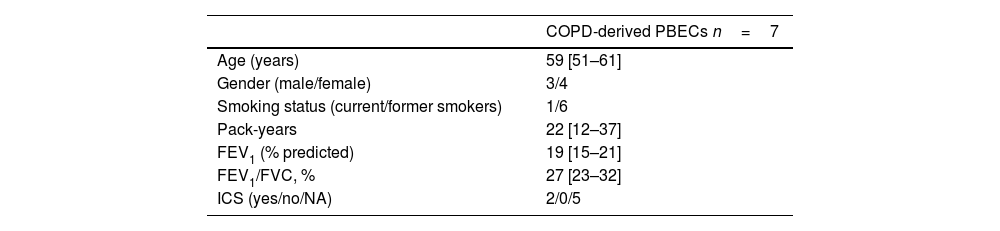
Material and methodsDonors of human primary bronchial epithelial cells (PBECs)PBECs were isolated by enzymatic treatment as described previously [15] from tracheobronchial tissue of transplanted lungs from 7 COPD patients with GOLD stage III-IV and from 7 non-COPD donor lungs. A full list of the COPD patient characteristics is provided in Table 1. No information was available on the lung donors. The study protocol was according to the research code of the UMCG (https://umcgresearch.org/w/research-code-umcg) and the national and ethical professional guidelines on the use of human material (https://www.coreon.org/wp-content/uploads/2023/06/Code-of-Conduct-for-Health-Research-2022.pdf).
Characteristics of COPD PBEC donors.
| COPD-derived PBECs n=7 | |
|---|---|
| Age (years) | 59 [51–61] |
| Gender (male/female) | 3/4 |
| Smoking status (current/former smokers) | 1/6 |
| Pack-years | 22 [12–37] |
| FEV1 (% predicted) | 19 [15–21] |
| FEV1/FVC, % | 27 [23–32] |
| ICS (yes/no/NA) | 2/0/5 |
Data are presented as median (IQR). PBECs: primary bronchial epithelial cells; FEV1: forced expiratory volume in 1second; FVC: forced vital capacity; ICS: inhaled corticosteroids; NA: not available.
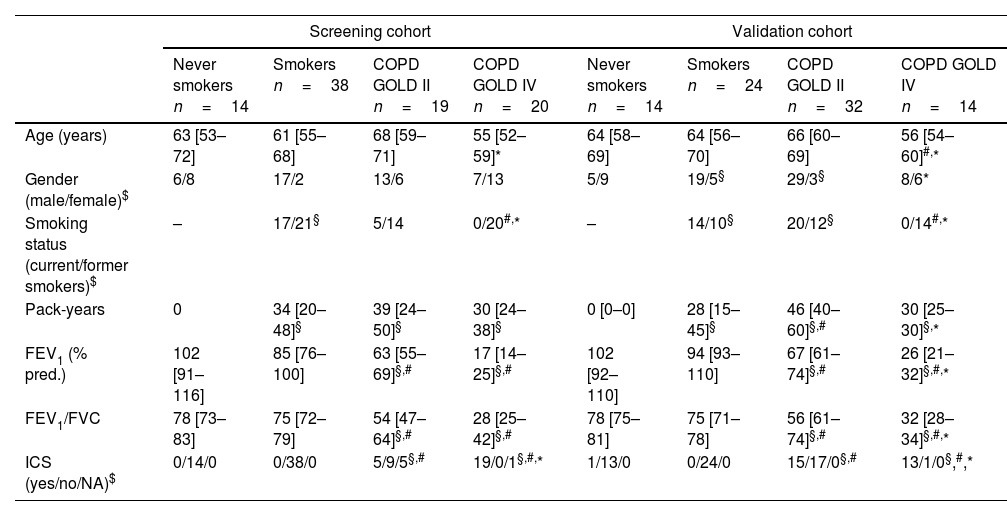
Peripheral lung tissue (containing parenchyma and small airways) was obtained from COPD patients with GOLD stage (III)–IV after lung transplantation or from non-COPD control donors and COPD patients with GOLD stage II after tumor resection surgery, taken as far as possible from the tumor and checked for abnormalities by an experienced pathologist. Patient characteristics are provided in Table 2 and Supplementary Tables S1 and S2. In the screening cohort, lung tissue was collected according to the research code of the University Medical Center Groningen (UMCG) and national ethical and professional guidelines. Sample collection of the validation cohort was approved by the medical ethics committee of Ghent University Hospital (2016/0132) and University Hospital Gasthuisberg, Leuven (S51577). Written informed consent of all subjects was provided.
Characteristics of lung tissue screening and validation cohort.
| Screening cohort | Validation cohort | |||||||
|---|---|---|---|---|---|---|---|---|
| Never smokers n=14 | Smokers n=38 | COPD GOLD II n=19 | COPD GOLD IV n=20 | Never smokers n=14 | Smokers n=24 | COPD GOLD II n=32 | COPD GOLD IV n=14 | |
| Age (years) | 63 [53–72] | 61 [55–68] | 68 [59–71] | 55 [52–59]* | 64 [58–69] | 64 [56–70] | 66 [60–69] | 56 [54–60]#,* |
| Gender (male/female)$ | 6/8 | 17/2 | 13/6 | 7/13 | 5/9 | 19/5§ | 29/3§ | 8/6* |
| Smoking status (current/former smokers)$ | – | 17/21§ | 5/14 | 0/20#,* | – | 14/10§ | 20/12§ | 0/14#,* |
| Pack-years | 0 | 34 [20–48]§ | 39 [24–50]§ | 30 [24–38]§ | 0 [0–0] | 28 [15–45]§ | 46 [40–60]§,# | 30 [25–30]§,* |
| FEV1 (% pred.) | 102 [91–116] | 85 [76–100] | 63 [55–69]§,# | 17 [14–25]§,# | 102 [92–110] | 94 [93–110] | 67 [61–74]§,# | 26 [21–32]§,#,* |
| FEV1/FVC | 78 [73–83] | 75 [72–79] | 54 [47–64]§,# | 28 [25–42]§,# | 78 [75–81] | 75 [71–78] | 56 [61–74]§,# | 32 [28–34]§,#,* |
| ICS (yes/no/NA)$ | 0/14/0 | 0/38/0 | 5/9/5§,# | 19/0/1§,#,* | 1/13/0 | 0/24/0 | 15/17/0§,# | 13/1/0§,#,* |
Data are presented as median with interquartile range. FEV1: forced expiratory volume in 1second; FVC: forced vital capacity; ICS: inhaled corticosteroids; NA: not available. Mann–Whitney U test was performed, unless otherwise described.
PBECs were stored in liquid nitrogen until use at passage 4 and cultured in bronchial epithelium growth medium (AEGM; Promocell, Heidelberg, Germany) and 1% Penicillin-Streptomycin (ThermoFisher) in collagen/fibronectin-coated flasks until confluence, as described previously [16].
Preparation of CSECSE was prepared as described previously [17]. In short, Kentucky 2R4F research cigarettes (The Tobacco Research Institute, University of Kentucky, Lexington, KY) were used without filter. Smoke from two cigarettes was bubbled through 25mL bronchial epithelial cell basal medium (100% CSE). The extract was prepared freshly and used within 15min.
Overexpression of miR-320d and CSE stimulation in PBECsPBECs from n=7 donors per group, based on statistically significant effects in previous studies [14,16], were seeded in duplicate in 500μl AEGM (PromoCell) at a density of 5×104cells/well in 24-wells plates. After the cells reached ∼75% confluence, they were transfected with 1nM pre-miRNA-precursor miR-320d (Qiagen, Hilden, Germany) or 1nM mimic control (Qiagen) using RNAimax (Invitrogen) in Gibco Opti-MEM (ThermoFisher) for 4h. After 4h, the transfection reagent was removed and cells were incubated in hormone/growth factor-deprived medium for 16h. Thereafter, cells were stimulated with 20% cigarette smoke extract (CSE) for 6 and 24h at 37°C 5% CO2. Cell-free supernatants were collected, cells were washed twice with PBS and harvested to isolate RNA and determine CXCL8, IL-1α and GM-CSF protein levels.
RNA extraction, quantification of miRNA expression and qPCRTotal RNA was extracted from the lung tissue samples of the screening cohort and from PBECs derived from non-COPD and COPD patients by TRIzol reagent method (Molecular Research Center, Cincinnati, OH) according to manufacturer's guidelines. RNA from the lung tissue samples of the validation cohort was extracted using the miRNeasy mini kit (Qiagen). Quantification of miRNA expression and qPCR was performed as described in the online data supplement.
GM-CSF and CXCL8 analysis by enzyme-linked immunosorbent assayThe levels of GM-CSF, IL-1α and CXCL8 were measured in cell-free supernatants from PBECs cultures using the human CXCL8, IL-1α and GM-CSF DuoSet ELISA kit (R&D Systems, Minneapolis, MN) according to manufacturer's protocol.
RNA sequencingLibrary preparation from total RNA isolated from non-COPD-derived PBECs transfected with miR-320d or mimic control and stimulated with or without CSE and RNA sequencing was performed as described in the online data supplement. For each sample, the trimmed reads were mapped to the human GRCh37.75 using Burrows–Wheeler Transform (STAR2 v2.5.4) and quantification was determined with HTSeq (v0.11.0).
Statistical analysisData were analyzed using the nonparametric rank sum Mann–Whitney U-test to determine significant differences between the different subject in the screening and validation cohort and the Wilcoxon signed-rank test to compare between different conditions in vitro. RNA sequencing data were analyzed using DESeq2 according to the design∼subject (version 2-1.14). Results were corrected for multiple testing with Benjamin–Hochberg false discovery rate (FDR), FDR-values lower than 0.05 were considered as statistically significant. Genes that were significant lower expressed after miR-320d overexpression were checked for predicted target genes according to TargetScan (v7.2) and miRDB (v6.0) [18–20].
Gene ontology and pathway enrichmentGenes with significant higher and lower expression after miR-320d overexpression were ranked based on significance and used to identify enrichment of biological processes and Reactome pathways according to g:Profiler (GO:BP and Reactome release 2021-04-01) [21].
ResultsOverexpression of miR-320d reduces CXCL8 and IL-1α levels in non-COPD-derived PBECs which is compromised in COPD-derived PBECsWe first investigated whether the suppressive effects of miR-320d overexpression on pro-inflammatory cytokine expression differ between non-COPD and COPD-derived PBECs. Overexpression of miR-320d mimic was confirmed by qPCR, resulting in an equally strong induction in both non-COPD and COPD-derived PBECs (Fig. 1a). Of note, miR-320d was not differentially expressed between COPD and control at baseline, but showed lower expression in response to CSE in COPD compared to control-derived cells, where CSE induced a significant upregulation of miR-320d (Supplementary Fig. 1a, b). In non-COPD-derived PBECs, CSE significantly increased CXCL8 and GM-CSF, but not IL-1α secretion. Overexpression of miR-320d significantly reduced secretion of all cytokines, both at baseline and after CSE stimulation (Fig. 1b–d). In COPD-derived PBECs, GM-CSF but not CXCL8 nor IL-1α secretion was significantly increased by CSE. Of note, IL-1α levels tended to be higher at baseline compared to non-COPD-derived PBECs (P=0.0728). Interestingly, miR-320d overexpression was unable to reduce CXCL8 and IL-1α secretion in COPD-derived PBECs, either at baseline and upon CSE exposure, with only a trend toward CXCL8 levels after CSE stimulation (Fig. 1b, c), and no effect at all on IL-1α release. The difference in effect of miR-320d mimic on IL-1α secretion between COPD-derived PBECs and cells from non-COPD controls was significant in the presence of CSE (Supplementary Fig. S2a). GM-CSF responses were similar in non-COPD and COPD-derived PBECs, and the suppressive effect of miR-320d overexpression on GM-GSF secretion was unaffected in COPD-derived PBECs (Fig. 1d).
 of non-COPD controls (n=7) and COPD patients (n=7) were grown to 70% confluence, transfected with 1nM miR-320d or mimic control for 4h, growth factor-deprived overnight and stimulated with 0% (baseline) or 20% cigarette smoke extract (CSE) for 24h. Cells were harvested for RNA isolation and cell-free supernatants were harvested to asses the pro-inflammatory cytokine levels. (a) miR-320d expression assessed by qPCR, normalized to housekeeping gene RNU48 and expressed as fold induction compared to mimic control (2−ΔΔCt). Medians with interquartile ranges are indicated. The baseline and CSE-induced (b) CXCL8, (c) IL-1α and (d) GM-CSF secretion upon overexpression of miR-320d or mimic control was determined in non-COPD-derived and COPD-derived PBECs. *P<0.05 between the indicated values as assessed by the Wilcoxon signed rank test for paired observations.")
Effects of miR-320d on pro-inflammatory cytokines in non-COPD and COPD-derived PBECs. Primary bronchial epithelial cells (PBECs) of non-COPD controls (n=7) and COPD patients (n=7) were grown to 70% confluence, transfected with 1nM miR-320d or mimic control for 4h, growth factor-deprived overnight and stimulated with 0% (baseline) or 20% cigarette smoke extract (CSE) for 24h. Cells were harvested for RNA isolation and cell-free supernatants were harvested to asses the pro-inflammatory cytokine levels. (a) miR-320d expression assessed by qPCR, normalized to housekeeping gene RNU48 and expressed as fold induction compared to mimic control (2−ΔΔCt). Medians with interquartile ranges are indicated. The baseline and CSE-induced (b) CXCL8, (c) IL-1α and (d) GM-CSF secretion upon overexpression of miR-320d or mimic control was determined in non-COPD-derived and COPD-derived PBECs. *P<0.05 between the indicated values as assessed by the Wilcoxon signed rank test for paired observations.
Next, we aimed to gain insight into the mechanisms of miR-320d regulatory effects on CSE-induced pro-inflammatory responses. Showing the strongest effects of miR-320d overexpression, gene expression profiles were assessed in control PBECs. At baseline, no genes were significantly altered in PBECs upon transfection with miR-320d mimic compared to mimic control (Fig. 2a). Upon CSE exposure, transfection with miR-320d resulted in differential expression of 137 genes compared to mimic control (Fig. 2b), of which 55 genes were significantly upregulated and 82 were significantly downregulated. See Supplementary Tables S3 and S4 for all differentially expressed genes and Supplementary Fig. S3 for plots from the top 5 most significant genes (CRCT1, KCNG1, ADAM8, CLCA4 and CST6). Of the 82 downregulated genes, 5 genes (ARHGAP31, C19ORF26, VIM, VPS37B and RAP2B) were predicted targets of miR-320d according to TargetScan or miRDB.
 of non-COPD controls (n=10) were grown to 70% confluence, transfected with 1nM miR-320d or mimic control for 4h, growth factor-deprived overnight and stimulated with 0% (baseline) or 20% cigarette smoke extract (CSE) for 24h. Cells were harvested for RNA isolation and gene expression profiles were determined by RNA sequencing and qPCR. (a) volcano plot of gene expression profiles in PBECs transfected with mimic control versus miR-320d mimic at baseline. (b) volcano plot of gene expression profiles in PBECs transfected with mimic control versus miR-320d mimic after 20% CSE stimulation. The top 5 gene names with the highest and lowest P-value were shown. In red the upregulated genes and in blue the downregulated genes. (c) IRAK1 expression was assessed by qPCR in non-COPD donors (n=6) and COPD donors (n=4), related to housekeeping genes B2M and PPIA and expressed as 2−ΔCt. (d) IRAK1 expression was assessed by qPCR in non-COPD (n=6) and COPD donors (n=4), related to housekeeping genes B2M and PPIA and expressed as fold induction compared to mimic control (2−ΔΔCt). *P<0.05 between the indicated values as assessed by the Wilcoxon signed rank test for paired observations within groups or the Mann–Whitney U test between groups. NS; not significant.")
Altered gene expression upon miR-320d overexpression in PBECs. Primary bronchial epithelial cells (PBECs) of non-COPD controls (n=10) were grown to 70% confluence, transfected with 1nM miR-320d or mimic control for 4h, growth factor-deprived overnight and stimulated with 0% (baseline) or 20% cigarette smoke extract (CSE) for 24h. Cells were harvested for RNA isolation and gene expression profiles were determined by RNA sequencing and qPCR. (a) volcano plot of gene expression profiles in PBECs transfected with mimic control versus miR-320d mimic at baseline. (b) volcano plot of gene expression profiles in PBECs transfected with mimic control versus miR-320d mimic after 20% CSE stimulation. The top 5 gene names with the highest and lowest P-value were shown. In red the upregulated genes and in blue the downregulated genes. (c) IRAK1 expression was assessed by qPCR in non-COPD donors (n=6) and COPD donors (n=4), related to housekeeping genes B2M and PPIA and expressed as 2−ΔCt. (d) IRAK1 expression was assessed by qPCR in non-COPD (n=6) and COPD donors (n=4), related to housekeeping genes B2M and PPIA and expressed as fold induction compared to mimic control (2−ΔΔCt). *P<0.05 between the indicated values as assessed by the Wilcoxon signed rank test for paired observations within groups or the Mann–Whitney U test between groups. NS; not significant.
To identify potential biological processes and pathways affected by miR-320d overexpression, we performed biological processes and pathway enrichment analyses in g:Profiler with either the significantly upregulated or downregulated genes by miR-320d overexpression in CSE-exposed PBECs. The higher expressed genes were enriched for “interleukin-33-mediated signaling pathway”. The top 3 enriched processes for lower expressed genes were “response to cytokine”, “cellular response to cytokine stimulus” and “protein targeting” and the top 3 reactome pathways were “eukaryotic translation elongation”, “late endosomal micro-autophagy” and “peptide chain elongation”. See Supplementary Table S5 for all enriched biological processes and reactome pathways.
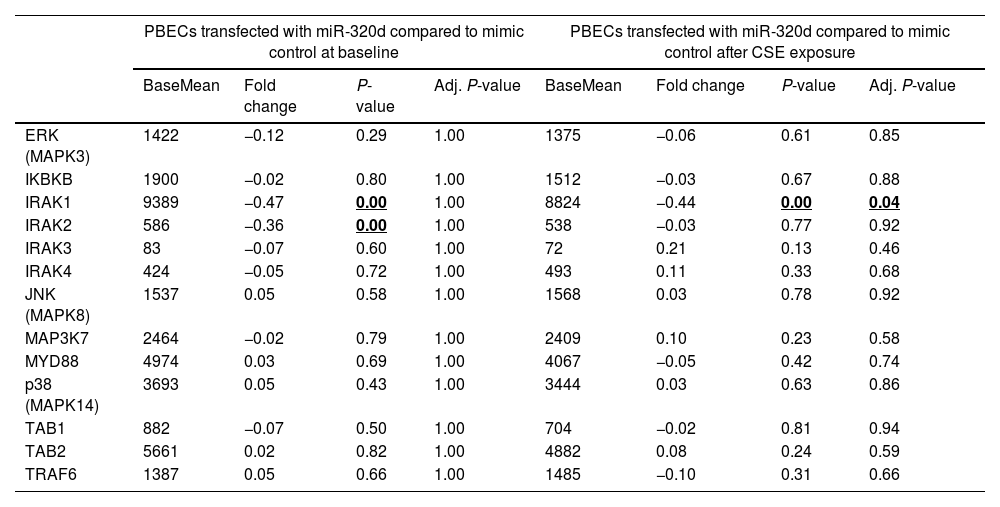
Differentially expressed genes upon miR-320d overexpression are involved in the IL-1β/NF-κB signalingSince we previously observed that miR-320d is able to reduce IL-1β-induced activation of the pro-inflammatory factor NF-κB [14], we specifically investigated expression of ERK, IKBKB, IRAK1-4, JNK, MAP3K7, MYD88, p38, TAB1, TAB2 and TRAF6, genes involved in this pathway [22]. Overexpression of miR-320d significantly downregulated IRAK1 expression after CSE exposure (Table 3; FDR adjusted P-value<0.05; Supplementary Fig. S4). The miR-320d-induced downregulation of IRAK1 was confirmed by qPCR, the effect being significant at baseline but not in the presence of CSE (Fig. 2c, d). While we did not observe significant differences in baseline IRAK1 expression between COPD and control in unexposed PBECs (Fig. 2c), miR-320d mimic was unable to suppress IRAK1 mRNA expression in COPD-derived PBECs, with a significant difference in effect between control and COPD-derived PBECs (Fig. 2d, Supplementary Fig. S2b).
Gene expression of genes involved in IL-1β-induced activation of NF-κB pathway.
| PBECs transfected with miR-320d compared to mimic control at baseline | PBECs transfected with miR-320d compared to mimic control after CSE exposure | |||||||
|---|---|---|---|---|---|---|---|---|
| BaseMean | Fold change | P-value | Adj. P-value | BaseMean | Fold change | P-value | Adj. P-value | |
| ERK (MAPK3) | 1422 | −0.12 | 0.29 | 1.00 | 1375 | −0.06 | 0.61 | 0.85 |
| IKBKB | 1900 | −0.02 | 0.80 | 1.00 | 1512 | −0.03 | 0.67 | 0.88 |
| IRAK1 | 9389 | −0.47 | 0.00 | 1.00 | 8824 | −0.44 | 0.00 | 0.04 |
| IRAK2 | 586 | −0.36 | 0.00 | 1.00 | 538 | −0.03 | 0.77 | 0.92 |
| IRAK3 | 83 | −0.07 | 0.60 | 1.00 | 72 | 0.21 | 0.13 | 0.46 |
| IRAK4 | 424 | −0.05 | 0.72 | 1.00 | 493 | 0.11 | 0.33 | 0.68 |
| JNK (MAPK8) | 1537 | 0.05 | 0.58 | 1.00 | 1568 | 0.03 | 0.78 | 0.92 |
| MAP3K7 | 2464 | −0.02 | 0.79 | 1.00 | 2409 | 0.10 | 0.23 | 0.58 |
| MYD88 | 4974 | 0.03 | 0.69 | 1.00 | 4067 | −0.05 | 0.42 | 0.74 |
| p38 (MAPK14) | 3693 | 0.05 | 0.43 | 1.00 | 3444 | 0.03 | 0.63 | 0.86 |
| TAB1 | 882 | −0.07 | 0.50 | 1.00 | 704 | −0.02 | 0.81 | 0.94 |
| TAB2 | 5661 | 0.02 | 0.82 | 1.00 | 4882 | 0.08 | 0.24 | 0.59 |
| TRAF6 | 1387 | 0.05 | 0.66 | 1.00 | 1485 | −0.10 | 0.31 | 0.66 |
PBECs: primary bronchial epithelial cells; CSE: cigarette smoke extract.
To evaluate whether miR-320d is differently expressed in COPD, its expression in whole lung tissue samples of COPD patients was compared to never smokers and (current/former) control smokers from independent screening (n=91) and validation (n=84) cohorts. Subject characteristics are shown in Table 1. In the screening cohort, we observed significantly lower miR-320d levels in control smokers and COPD patients compared to never smokers (Fig. 3a). In the validation cohort, lower miR-320d levels in COPD patients compared to never smokers were confirmed (Fig. 3b). Assessing the impact of disease severity on miR-320d expression, we observed no significant differences between COPD patients with GOLD stage II or IV and control smokers in the screening cohort (Fig. 3c). In the validation cohort, miR-320d levels were significantly lower in COPD patients with GOLD stage IV compared to those with GOLD stage II and control smokers (Fig. 3d). In line with the lower expression of miR-320d in control and COPD (ex-)smokers, we observed a significant negative correlation between miR-320d expression and packyears in the screening, but not validation cohort (Supplementary Fig. S5). In line with the lower expression of miR-320d with COPD severity in the validation cohort, we observed a significant positive correlation between miR-320d expression and FEV1 as well as diffusing capacity of the lungs for carbon monoxide (DLCO) in the validation, but not screening cohort (Supplementary Fig. S5). These results demonstrate miR-320d expression is lower in smokers and may be further reduced with COPD severity.
 and expressed as 2−ΔCt. (a) Comparison of never smokers, control smokers and COPD patients in the screening cohort. (b) Comparison of never smokers, control smokers and COPD patients in the validation cohort. (c) Comparison of smoking controls, COPD GOLD II and COPD GOLD IV patients in the screening cohort. (d) Comparison of smoking controls, COPD GOLD II and COPD GOLD III–IV patients in the validation cohort. Medians with interquartile range are indicated. The closed triangles indicate control current smokers and open triangles indicate control former smokers. The open circles indicated COPD patients with GOLD II and closed circles indicated COPD patients with GOLD (III)–IV. *P<0.05, ***P<0.001, ****P<0.0001 between the indicated values as analyzed with the Mann–Whitney U test.")
miR-320d expression in lung tissue of never/former/current smoking non-COPD control subjects and patients with GOLD stage II–IV COPD. Lung tissue was derived from never, current and former smoking controls and COPD patients. The expression of miR-320d was measured in lung tissue by qPCR. Expression of miR-320d was normalized to housekeeping genes (RNU44 and RNU48) and expressed as 2−ΔCt. (a) Comparison of never smokers, control smokers and COPD patients in the screening cohort. (b) Comparison of never smokers, control smokers and COPD patients in the validation cohort. (c) Comparison of smoking controls, COPD GOLD II and COPD GOLD IV patients in the screening cohort. (d) Comparison of smoking controls, COPD GOLD II and COPD GOLD III–IV patients in the validation cohort. Medians with interquartile range are indicated. The closed triangles indicate control current smokers and open triangles indicate control former smokers. The open circles indicated COPD patients with GOLD II and closed circles indicated COPD patients with GOLD (III)–IV. *P<0.05, ***P<0.001, ****P<0.0001 between the indicated values as analyzed with the Mann–Whitney U test.
In this study, we confirmed the anti-inflammatory effect of miR-320d, suppressing baseline and CSE-induced pro-inflammatory responses in bronchial epithelial cells, and we show that this effect is attenuated in COPD. RNA-sequencing revealed that miR-320d overexpression particularly affected gene expression profiles upon CSE exposure. The genes downregulated by miR-320d were associated with the pathway “response to cytokines”, which supports the notion that miR-320d is involved in the suppression of pro-inflammatory responses in cigarette smoke-exposed airway epithelium. Within this pathway, miR-320d overexpression significantly downregulated gene expression of IRAK1 in non-COPD PBECs. Importantly, this downregulation of IRAK1 did not occur in COPD-derived PBECs, with miR-320d being unable to inhibit the secretion of pro-inflammatory cytokines CXCL8 and IL-1α. Finally, we observed lower expression of miR-320d in peripheral lung tissue of smokers and COPD patients compared to never smokers, with the lowest expression in severe COPD patients in the validation cohort. These data suggest that the anti-inflammatory action of miR320d could be impaired in smokers and COPD patients due to lower expression levels and/or reduced effectiveness.
CSE exposure increased CXCL8 and GM-CSF secretion in non-COPD-derived PBECs, while no significant effect on IL-1α was observed. This is not fully in accordance with our previous study were we observed higher IL-1α levels upon CSE exposure [23], which could be due to slight differences in the experimental setup. Unexpectedly, we did not observe a stronger CSE-induced increase in pro-inflammatory mediators in COPD-derived PBECs. This may in part be due to the higher baseline secretion of these cytokines in some of the COPD-derived cultures. Notably, overexpression of miR-320d reduced GM-CSF, IL-1α and CXCL8 secretion in non-COPD-derived PBECs both at baseline and after CSE exposure, while in COPD-derived PBECs, miR-320d overexpression did not reduce IL-1α and CXCL8 secretion neither at baseline nor after CSE exposure. Therefore, we suggest that this reduced effectiveness of miR-320d may contribute to the abnormal inflammatory responses in COPD. In future studies, it will be of interest to investigate whether this compromised anti-inflammatory response upon miR-320d upregulation may be related to ICS insensitivity in COPD. Given the finding that miR-320d upregulated genes involved in IL-33-mediated signaling, it may additionally be of interest to assess whether differences in miR-320d expression may be related to heterogenity in responsiveness to biologicals.
When assessing the effect of miR-320d on the transcriptional response, we identified CLCA4 as most upregulated and CRCT1 as most repressed gene. CLCA4 encodes a channel involved in chloride transport into epithelial cells [24], while CRCT1 encodes the cysteine rich C-terminal 1 protein that may be involved in keratinocyte differentiation [25], yet little is known regarding the expression of CLCA4 and CRCT1 in COPD and their involvement in inflammatory pathways. The gene expression profiles further revealed that lower expressed genes were most significantly associated with the biological process “response to cytokine”. Interleukin 1 receptor associated kinase 1 (IRAK1) is a gene involved in this biological process that can be activated by cigarette smoke [26] and that was suppressed by miR-320d overexpression. Upon activation of the IL-1 receptor, IRAK1 forms a complex with MyD88 and TRAF6 to induce innate immune responses by activation of the NF-κB pathway [27], and may thus be involved in secretion of pro-inflammatory cytokines. Whether COPD is associated with genetic variants in the IRAK1 locus is unknown, and our findings remain to be confirmed at protein level. Nevertheless, our findings are supportive of abnormalities in the regulation of IRAK1, as the effect of miR-320d overexpression was significantly attenuated in COPD-derived epithelial cells, in line with the lack of suppressive effect on IL-1α secretion. Furthermore, 5 miR-320d-validated target genes (ARHGAP31, C19ORF26, VIM, VPS37B and RAP2B) were downregulated by miR-320d overexpression. To the best of our knowledge, the involvement of these genes in (aberrant) pro-inflammatory responses has not been studied in detail.
In addition to abnormalities in its regulatory effects in bronchial epithelium in vitro, we were interested in lung tissue expression of miR-320d in relation to COPD severity. In the validation cohort, miR-320d was significantly lower expressed in severe COPD patients compared to never smokers, control smokers and mild-moderate COPD patients. In the screening cohort, miR-320d levels were also lower in lung tissue of COPD patients compared to never smokers. However, no significant differences were observed between severe and mild/moderate COPD patients or non-COPD smokers. Possible explanations for the difference between the cohorts may be the heterogeneity of the disease or differences in the use of ICS between the cohorts, as ICS can upregulate miR-320d expression in COPD patients [14]. Although severe COPD patients in both cohorts used ICS, no information was available on the frequency and dosage of ICS use. Another discrepancy between the cohorts is the lower expression of miR-320d in control smokers versus never smokers in the screening but not validation cohort. An explanation may be the lower pack-years of the control smokers in the validation cohort, although this was just a trend. Since the expression of miR-320d was not different between males and females, the gender imbalance between the cohorts not likely contributes to the observed discrepancies.
Previously, slightly higher expression levels of miR-320d have been observed in lung tissue of smokers with compared to smokers without COPD [11]. No information on corticosteroid treatment was available, yet this may have contributed to the discrepancy with the observations in our study, as it was previously observed that ICS use upregulates miR-320d expression [14]. Besides the limitation of lacking information on the frequency and dosage of ICS use in the lung tissue cohorts, another limitation of our study is that ICS use may have contributed to the heterogeneity in responses in vitro, even though cells had been cultured for ∼2–3 weeks before experimentation. In addition, the sample size in the in vitro study was relatively small. Finally, we studied miR-320d expression in peripheral lung tissue containing parenchymal tissue as well as small airways, however no data were available on the actual number of airways present in the tissue. This limits the translation of these findings to the in vitro data on bronchial epithelial cells. Still, our results from the different compartments support our hypothesis on the dysregulation of miR-320d in COPD, in a translational study using unique cells from well-characterized COPD patients.
In summary, our study provides novel insight into the potential molecular mechanisms of persistent airway inflammation in COPD. It suggests that the anti-inflammatory effect of miR-320d on smoke-exposed bronchial epithelial pro-inflammatory responses may be compromised in COPD patients. Our data also indicate that variation in miR-320d expression may contribute to the heterogeneity in responsiveness of COPD patients to ICS as well as to novel biologicals. Thus, miR-320d may serve as a clinical biomarker in the future. We observed lower miR-320d expression in peripheral lung tissue of smoking controls and COPD patients compared to never smokers, with a further reduction in severe COPD patients. Thus, altered expression and/or action of miR-320d may contribute to the abnormal, sustained inflammation in COPD. Further functional studies are warranted to investigate the involvement of miR-320d in heterogeneity in the responsiveness to ICS as well as responsiveness to biologicals in COPD.
Author contributionsIHH, CAB, MvdB, and MPR conceived the project and designed the experiments; MPR, MRJ and KRB conducted the experiments and IHH supervised the practical work. All authors contributed to the data analysis and interpretation; MPR drafted the manuscript. IHH, CAB, TM and KRB critically reviewed the manuscript. All authors carefully read and edited the manuscript. All authors have approved the final version of the paper.
Artificial intelligence involvementNo artificial intelligence was used.
FundingThis project was funded by Stichting Asthma Bestrijding (SAB)2017/040. MR was funded by the U4 project (Ghent University and University Medical Centre Groningen, BOF05U40516).
Conflict of interestThe authors declare not to have any conflicts of interest that may be considered to influence directly or indirectly the content of the manuscript.
The authors would like to thank Prof. Wim Janssens, Dr. Bart Vanaudenaerde and Dr. Stijn Verleden (Department of Pneumology, Leuven) for providing us with the explant lungs of patients with severe COPD.
The followings are the supplementary data to this article:







