



Chronic obstructive pulmonary disease (COPD) and lung cancer (LC) frequently coexist. Smokers with airflow limitation face a six-fold increased LC risk compared to smokers with normal lung function, indicating an increased risk independent of smoking [1]. Approximately 50–70% of smokers with pre-existing COPD are diagnosed with non-small-cell lung cancer (NSCLC) [2], which accounts for about 80% of LC cases. This strong association, particularly with emphysema [3], has prompted investigations into shared pathological mechanisms, including oxidative stress, genetic predisposition, and altered immune responses. The chronic airway inflammation of COPD creates an immunosuppressive microenvironment that can contribute to LC development. T-cell inhibitory pathways, such as those involving cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1), critically modulate immune responses [4,5], and their overexpression can promote neoplastic development. Polymorphisms in their respective genes have been linked to cancer susceptibility, with CTLA-4 overexpression observed in COPD patients [6] and the PD-1/PD-L1 axis implicated in COPD pathogenesis [7]. Although immune checkpoint inhibitors show promise in NSCLC treatment, low response rates [8] highlight the need to understand other contributing factors. Interleukin (IL)-9 and IL-21, with their complex roles in inflammation and anti-tumor immunity [9,10], also have gene polymorphisms associated with various cancers [11,12]. Given the limited data on the impact of single nucleotide polymorphisms (SNPs) in these immune-related genes on LC predisposition in COPD subjects, especially those with emphysema, this exploratory study aimed to investigate the association between genetic polymorphisms in the CTLA4, PDCD1, IL9, and IL21 genes and LC susceptibility in patients with COPD.
Patients were recruited and categorized into four groups: COPD without emphysema (COPD-no-E), COPD with emphysema (COPD-E), COPD with emphysema and lung cancer (COPD-E-LC), and a control group of current and former smokers (CS) without airflow limitation. All participants underwent spirometry and received a low-density computed tomography (CT) scan as part of a LC screening program. COPD was diagnosed in patients with smoking history of ≥10pack/year and forced expiratory volume in the first second to forced vital capacity ratio (FEV1/FVC) less than 0.7. Exclusions included recent COPD exacerbations or other malignancies. Emphysema was diagnosed using high-resolution chest CT scans and a carbon monoxide diffusion capacity (DLCO)<80% predicted. LC was histologically confirmed in all COPD-E-LC patients. Genotyping of selected polymorphisms in CTLA4, PDCD1, IL9, and IL21 genes was performed in DNA extracted from blood nucleated cells using amplicon-based next-generation sequencing (NGS) on an Illumina MiSeq platform (online supplemental Table S1) following standard protocols. Statistical analyses involved Chi-squared tests, two-tailed Fisher's exact tests, or the Freeman–Halton extension to compare genotypic frequencies. Odds ratios (OR) and 95% confidence intervals (CI) were calculated across five genetic models (homozygous, heterozygous, dominant, over-dominant, and recessive). The genetic inheritance model for each polymorphism was chosen based on the lowest Akaike Information Criterion value (Supplemental Methods). Given the exploratory nature of this pilot study and its small sample size, OR values of ≥2 or ≤0.5 were considered indicative of a clinically relevant effect, with statistical significance set at p≤0.05.
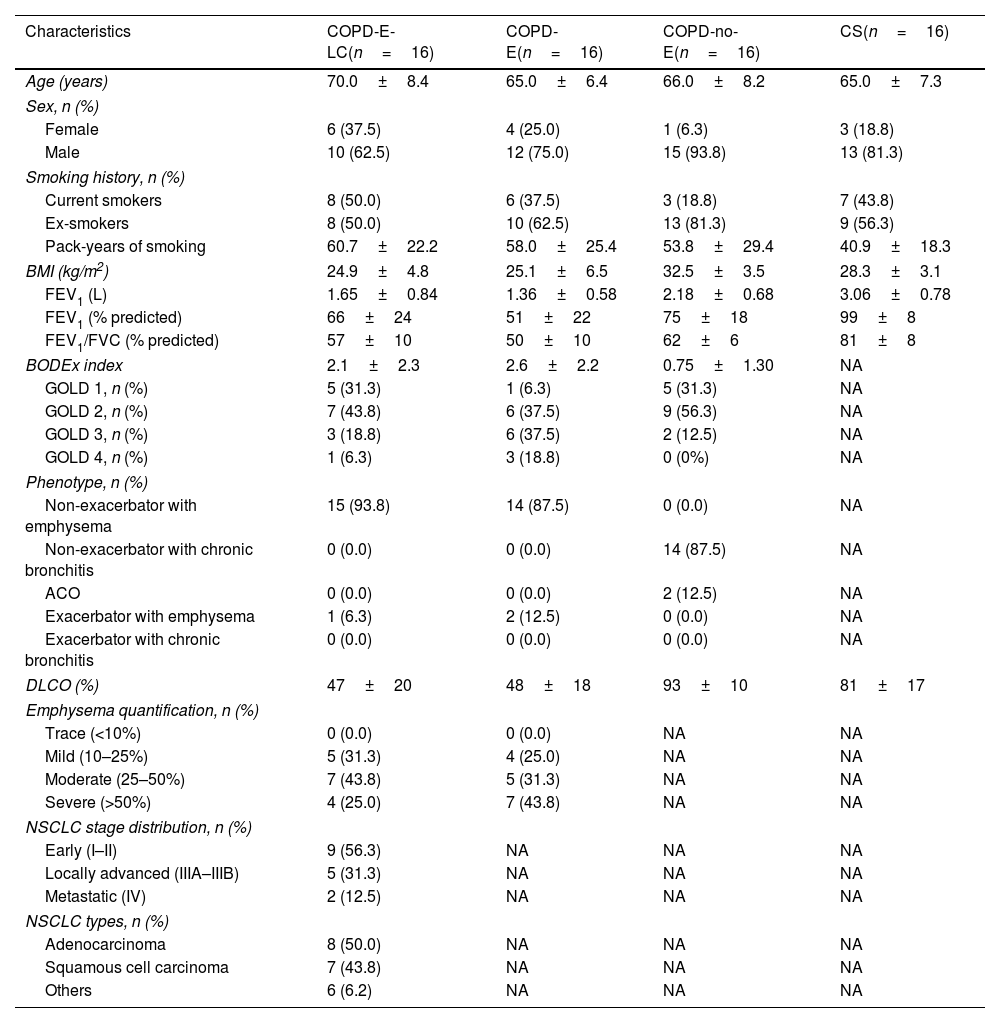
A total of 64 subjects were included in the study (Table 1). The COPD-no-E group had a significantly higher body mass index (BMI) (p<0.001) compared to the emphysematous COPD groups, which conversely showed more severe disease, as indicated by lower predicted FEV1%, lower DLCO%, and higher BODEx index scores (p=0.01, p<0.001, and p=0.03, respectively). Four of the 32 SNPs deviated from Hardy-Weinberg equilibrium (online supplemental Table S2) and were excluded from the analysis. Our investigation into genetic associations revealed that within the CTLA4 gene (Fig. 1, online supplemental Table S3) the rs11571315 polymorphism, under the heterozygous model (OR=0.250, 95% CI=0.05–1.25, p=0.091), and the rs231775 polymorphism under an over-dominant model (OR=0.218, 95% CI=0.047–1.005, p=0.05), showed a lower risk trend for developing COPD-E-LC in the CS group. This suggests that individuals with the CT genotype or who are carriers of the G allele, respectively, have a lower risk. Conversely, rs11571316 was associated with a higher risk trend of E-LC in the COPD-no-E group (OR=2.139, 95% CI=0.472–9.699), though it did not reach the required statistical significance (p=0.269).
Demographic and clinical characteristics of the four groups under study.
| Characteristics | COPD-E-LC(n=16) | COPD-E(n=16) | COPD-no-E(n=16) | CS(n=16) |
|---|---|---|---|---|
| Age (years) | 70.0±8.4 | 65.0±6.4 | 66.0±8.2 | 65.0±7.3 |
| Sex, n (%) | ||||
| Female | 6 (37.5) | 4 (25.0) | 1 (6.3) | 3 (18.8) |
| Male | 10 (62.5) | 12 (75.0) | 15 (93.8) | 13 (81.3) |
| Smoking history, n (%) | ||||
| Current smokers | 8 (50.0) | 6 (37.5) | 3 (18.8) | 7 (43.8) |
| Ex-smokers | 8 (50.0) | 10 (62.5) | 13 (81.3) | 9 (56.3) |
| Pack-years of smoking | 60.7±22.2 | 58.0±25.4 | 53.8±29.4 | 40.9±18.3 |
| BMI (kg/m2) | 24.9±4.8 | 25.1±6.5 | 32.5±3.5 | 28.3±3.1 |
| FEV1 (L) | 1.65±0.84 | 1.36±0.58 | 2.18±0.68 | 3.06±0.78 |
| FEV1 (% predicted) | 66±24 | 51±22 | 75±18 | 99±8 |
| FEV1/FVC (% predicted) | 57±10 | 50±10 | 62±6 | 81±8 |
| BODEx index | 2.1±2.3 | 2.6±2.2 | 0.75±1.30 | NA |
| GOLD 1, n (%) | 5 (31.3) | 1 (6.3) | 5 (31.3) | NA |
| GOLD 2, n (%) | 7 (43.8) | 6 (37.5) | 9 (56.3) | NA |
| GOLD 3, n (%) | 3 (18.8) | 6 (37.5) | 2 (12.5) | NA |
| GOLD 4, n (%) | 1 (6.3) | 3 (18.8) | 0 (0%) | NA |
| Phenotype, n (%) | ||||
| Non-exacerbator with emphysema | 15 (93.8) | 14 (87.5) | 0 (0.0) | NA |
| Non-exacerbator with chronic bronchitis | 0 (0.0) | 0 (0.0) | 14 (87.5) | NA |
| ACO | 0 (0.0) | 0 (0.0) | 2 (12.5) | NA |
| Exacerbator with emphysema | 1 (6.3) | 2 (12.5) | 0 (0.0) | NA |
| Exacerbator with chronic bronchitis | 0 (0.0) | 0 (0.0) | 0 (0.0) | NA |
| DLCO (%) | 47±20 | 48±18 | 93±10 | 81±17 |
| Emphysema quantification, n (%) | ||||
| Trace (<10%) | 0 (0.0) | 0 (0.0) | NA | NA |
| Mild (10–25%) | 5 (31.3) | 4 (25.0) | NA | NA |
| Moderate (25–50%) | 7 (43.8) | 5 (31.3) | NA | NA |
| Severe (>50%) | 4 (25.0) | 7 (43.8) | NA | NA |
| NSCLC stage distribution, n (%) | ||||
| Early (I–II) | 9 (56.3) | NA | NA | NA |
| Locally advanced (IIIA–IIIB) | 5 (31.3) | NA | NA | NA |
| Metastatic (IV) | 2 (12.5) | NA | NA | NA |
| NSCLC types, n (%) | ||||
| Adenocarcinoma | 8 (50.0) | NA | NA | NA |
| Squamous cell carcinoma | 7 (43.8) | NA | NA | NA |
| Others | 6 (6.2) | NA | NA | NA |
COPD-E-LC, patients with COPD, emphysema and lung cancer; COPD-E, patients with COPD and emphysema; COPD-no-E, patients with COPD without emphysema; CS, control group of current and former smokers; BMI, body mass index; BODEx, Body mass index (B), Obstruction (O), Dyspnea (D) and Exacerbations of COPD (Ex). FEV1, forced expiratory volume in 1s; FVC, forced vital capacity; ACO, asthma-COPD overlap; DLCO, diffusion lung capacity; NA, non-applicable.
 and CI 95% for the studied polymorphisms are shown. COPD-E-LC, patients with COPD, emphysema and lung cancer; COPD-E, patients with COPD and emphysema; COPD-no-E, patients with COPD without emphysema; CS, control group of current and former smokers.")
Forest plots of polymorphisms in the CTLA4, PDCD1 and IL9 genes and lung cancer risk in study groups. Odds ratios (ORs) and CI 95% for the studied polymorphisms are shown. COPD-E-LC, patients with COPD, emphysema and lung cancer; COPD-E, patients with COPD and emphysema; COPD-no-E, patients with COPD without emphysema; CS, control group of current and former smokers.
Regarding the PDCD1 gene (Fig. 1, online supplemental Table S3), rs11568821 was associated with a higher risk trend of LC in the COPD-E group (OR=2.800, 95% CI=0.427–18.38), while rs2227981 was linked to a higher risk trend of developing COPD-E-LC in the CS group but a lower risk trend of LC in both COPD groups, suggesting differential genetic effects depending on COPD and emphysema status, though none of these associations reached statistical significance. Further analysis of the IL9 gene revealed several significant associations (Fig. 1, online supplemental Table S4). The rs2069870 A allele was associated with a significantly increased risk trend of LC in the overall COPD group due to its larger sample size (OR=3.792, 95% CI=0.985–14.59, p=0.05). Another polymorphism, rs1859430 showed a significant association with an increased risk trend of LC in the overall COPD group under a dominant model (OR=3.792, 95% CI=0.985–14.59, p=0.05), suggesting that G allele carriers have a higher susceptibility. Conversely, the rs31564 polymorphism was significantly associated with a lower LC risk in the overall COPD group and a lower risk trend in the COPD-E group for the heterozygous TG genotype but showed a higher risk trend in the COPD-no-E group. On the other hand, the polymorphisms rs2069882 and rs2069885 in the COPD-no-E group, and rs11741137 in the COPD-no-E and CS groups showed suggestive, but non-significant, associations with a higher risk trend.
For the IL21 gene, the rs907715 polymorphism, under a dominant model, showed a non-significant trend for an increased risk of LC for T allele carriers in COPD groups (online supplemental Table S4).
The results of our study showed that the CTLA4 rs231775 G allele conferred a lower risk trend of COPD-E-LC in CS group. This finding aligns with previous research suggesting the A allele increases LC risk [13], and is linked to shorter survival times compared to the GG genotype [14]. Mechanistically, the Ala (GG) for Thr (AA) substitution enhances the CTLA-4/B7.1 interaction, which leads to greater T cell inhibition and reduced IL-2 production [13], whilst the GG genotype promotes higher T lymphocyte proliferation than the AA genotype [15]. However, the absence of this association in our COPD groups is intriguing. Given the similar genotypic and allelic frequencies between COPD-E-LC and COPD groups, our results strongly suggest that the underlying genetic variations prevalent in COPD might inherently contribute to the heightened LC susceptibility observed in COPD patients, potentially masking individual SNP effects when compared only within COPD cohorts. The controversial impact of rs11571315 on CTLA-4 transcription [16], and our observed lower risk trend of COPD-E-LC in CS for heterozygous individuals warrants further investigation into shared genetic variations and their implications for CTLA-4 expression in the context of COPD.
The PD-1/PD-L1 pathway is a key target for immunotherapy in cancers like NSCLC, and patients who also have COPD have shown better treatment outcomes [17]. While previous research has linked the PDCD1 polymorphism rs2227981 to a reduced risk of LC [18], our study did not find a significant association between several PDCD1 polymorphisms, including rs2227981, and the risk of COPD-E-LC in the CS group. However, the findings suggest that COPD, with or without emphysema might modify the risk of developing LC. For instance, COPD-E patients with heterozygous rs11568821 genotype may exhibit increased susceptibility, whereas COPD carriers of the A allele in rs2227981 may have a reduced risk trend, contrasting with the increased risk trend of COPD-E-LC observed in the CS group. Further research is needed to validate these potential associations.
Polymorphisms in the IL9 gene may play a role in LC susceptibility in individuals with COPD. Prior research has shown that IL-9 can contribute to an immunosuppressive tumor microenvironment [19] and that its deficiency may reduce tumor burden [9], underscoring its dual and complex role. Our study found that the IL9 rs2069870 A allele, in conjunction with rs1859430, was associated with a significantly increased risk trend of LC in the overall COPD group, demonstrating a large effect size (OR=3.792). Furthermore, our results suggest that the influence of emphysema is complex, as evidenced by the contrasting effects of the rs31564 polymorphism on LC risk between COPD patients with and without emphysema Finally, our data strongly suggest that COPD-no-E individuals who are heterozygous for rs2069882, rs2069885 and rs11741137 may have an increased risk trend of developing E-LC, a finding that is consistent with the increased risk trend observed in heterozygous individuals for rs11741137 within the CS group. Similarly, associations of IL21 rs907715 with an increased LC risk trend in both COPD groups suggest that genetic variations affecting this anti-tumor cytokine may also contribute to susceptibility.
This exploratory pilot study, despite its limited sample size, provides compelling preliminary evidence of a genetic link between LC and COPD, critically highlighting that the presence of emphysema significantly alters the impact of specific SNPs on LC risk. Our findings indicate that polymorphisms in CTLA4 (notably rs231775, rs7333618, rs11571316), IL9 (rs2069870, rs1859430, rs31564), and IL21 (rs907715) are promising for further research and potential prospective evaluation of LC risk in smokers, both with and without COPD enabling early interventions in LC screening. Nevertheless, validating these findings in larger cohorts remains essential.
Contribution of each authorPOG and JSC recruited the study participants and conducted pulmonary function tests. MDM and CGB performed NGS techniques. Statistical analysis: MDM, CGB, POG and JJSC. MDM and JJSC designed the study and drafted the initial manuscript. All authors contributed to data interpretation, revision of the manuscript, and approved the final version for submission.
Artificial intelligence involvementNone of the materials has been partially or totally produced with help of any artificial intelligence software or tool.
Funding of the researchThe present work was supported by the Valencian Society of Pulmonology (SVN)/Pulmonary Foundation of the Valencian Community (Grant UGP-19-483) to JJ Soler-Cataluña.
Conflicts of interestPOG, MDM and CGB have declared no conflicts of interest. JSC has received honoraria for lectures from Aflofarm, expert testimony fees from Astra Zeneca, and support for attending meetings from Boehringer Ingelheim. JJSC has received grants from GSK, meeting support from AstraZeneca, Chiesi, FAES and Menarini as well as consulting or speaking fees from AstraZeneca, Chiesi, GSK, Sanofi, FAES, Boehringer Ingelheim, Grifols, Menarini, MSD, and Zambon.
The authors are grateful for the kind and selfless collaboration of the study participants.
The followings are the supplementary data to this article:







