








La pirfenidona fue el primer fármaco antifibrótico aprobado en Argentina para fibrosis pulmonar idiopática. Los resultados de los ensayos clínicos podrían ser diferentes a los de la vida real. El objetivo primario fue estudiar la tolerancia de la pirfenidona en la vida real. El objetivo secundario analizar la eficacia y los motivos de suspensión.
Materiales y métodosEstudio observacional retrospectivo realizado en 4 centros especializados de Argentina. Se analizaron las historias clínicas de pacientes con fibrosis pulmonar idiopática que recibieron pirfenidona entre junio de 2013 y setiembre de 2016. Se analizaron efectos adversos y las variables que podrían influir en la ocurrencia de los mismos. Se comparó además la evolución de capacidad vital forzada (CVF%) entre los periodos prepirfenidona y pospirfenidona.
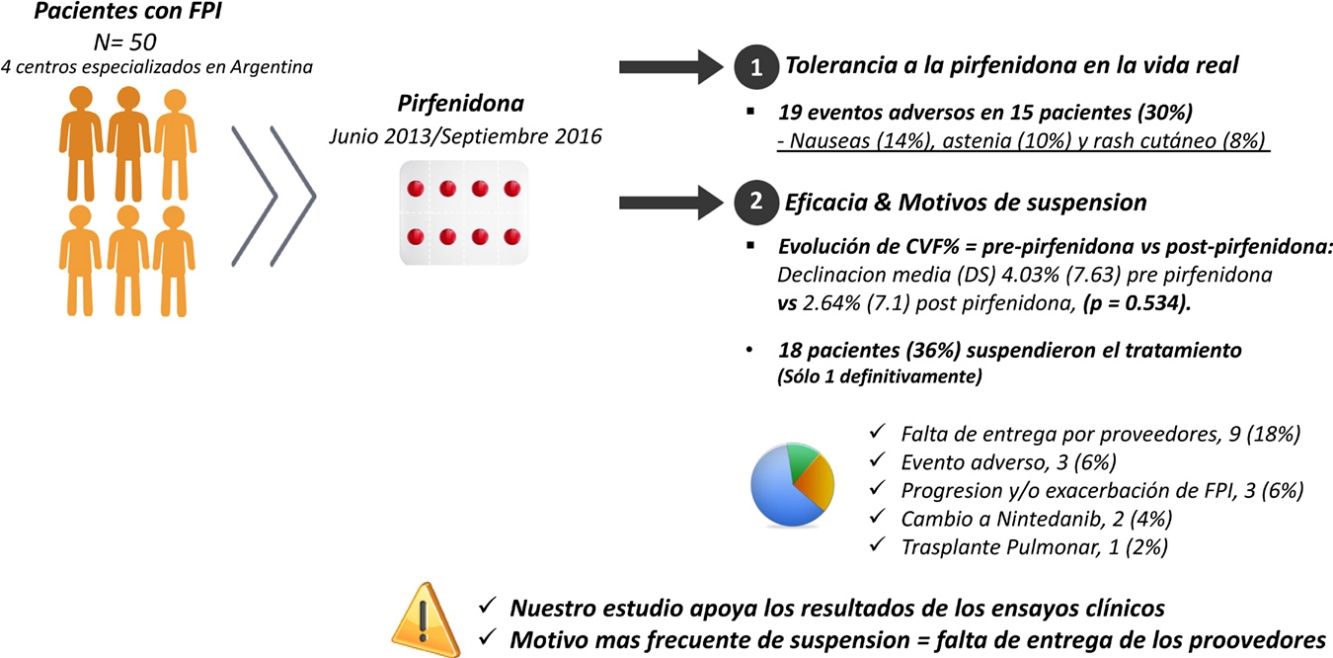
ResultadosCincuenta pacientes, 38 (76%) hombres, edad media (DE) 67,8 (8,36) años. La media (DE) de exposición a pirfenidona fue 645,68 (428,19) días, con una dosis diaria media (DE) de 2.064,56mg (301,49). Se reportaron 19 eventos adversos en 15 pacientes (30%): náuseas (14%), astenia (10%) y rash cutáneo (8%). Dieciocho pacientes (36%) interrumpieron el tratamiento, uno definitivamente. El motivo más frecuente fue la falta de entrega de proovedores en 9 (18%). Comparamos la evolución de CVF% entre los períodos prepirfenidona y pospirfenidona, con una declinación media (DE) de CVF% de 4,03% (7,63) prepirfenidona y 2,64% (7,1) pospirfenidona, (p=0,534).
ConclusionesEn nuestro estudio la pirfenidona fue bien tolerada y ha demostrado un enlentecimiento en la declinación de la CVF, aunque sin alcanzar significación estadística.
Pirfenidone was the first antifibrotic drug approved in Argentina for idiopathic pulmonary fibrosis (IPF). Outcomes in real life may differ from the results of clinical trials. The primary endpoint was to study the tolerance of pirfenidone in real life. Secondary endpoints were to analyze effectiveness and reasons for discontinuation.
Materials and methodsRetrospective observational study conducted in 4 specialized centers in Argentina. We analyzed the medical records of patients with IPF who received pirfenidone between June 2013 and September 2016. Adverse events (AE) and the variables that could influence these results were analyzed. Forced vital capacity (FVC%) parameters were also compared between the pre-pirfenidone and post-pirfenidone periods.
ResultsFifty patients were included, 38 (76%) men, with mean age (SD) 67.8 (8.36) years. Mean (SD) exposure to pirfenidone was 645.68 (428.19) days, with a mean daily dose (SD) of 2,064.56mg (301.49). Nineteen AEs in 15 patients (30%) were reported: nausea (14%), asthenia (10%) and skin rash (8%). A total of 18 patients (36%) interrupted treatment, only 1 definitively. The most frequent reason for discontinuation was failure of suppliers to provide the drug (9 subjects; 18%). We compared the evolution of FVC% between the pre-pirfenidone and post-pirfenidone periods, and found a mean (SD) FVC% decline of 4.03% (7.63) pre-pirfenidone and 2.64% (7.1) post-pirfenidone (P=.534).
ConclusionsIn our study, pirfenidone was well tolerated and associated with a reduction in FVC decline, although without reaching statistical significance.
La fibrosis pulmonar idiopática (FPI) es una enfermedad progresiva e irreversible que lleva en la mayoría de los casos a la muerte en 3 a 5 años1. En el año 2008 la pirfenidona se convirtió en el primer fármaco antifibrótico específico aprobado para el tratamiento de dicha enfermedad, y en el año 2012 fue aprobada en Argentina para su uso en pacientes con FPI leve a moderada2. Dicha aprobación se sostuvo en los resultados del ensayo clínico ASCEND, que confirmó una reducción en la declinación de la capacidad vital forzada (CVF) y una mejoría en la sobrevida libre de progresión y la tolerancia al ejercicio3. Si bien la eficacia y seguridad de la pirfenidona fue estudiada en 5 ensayos clínicos aleatorizados, doble ciego y controlados con placebo, los pacientes de dichos ensayos cumplían con estrictos criterios de inclusión y exclusión, motivo por el cual podrían no representar la heterogénea población de pacientes con FPI atendida en la práctica diaria2–6. Se han desarrollado diversos estudios de vida real, de manera que contamos actualmente con evidencia sobre su seguridad y eficacia por más de 6 años7–9. El perfil de seguridad y la eficacia de la pirfenidona pueden variar en las distintas poblaciones debido a diferencias en cuanto a polimorfismos genéticos, condiciones climáticas, costumbres alimenticias, entre otras. Esto subraya la importancia de la realización de estudios de vida real en distintos países. En Argentina desconocemos cuál es la frecuencia de los eventos adversos (EA), su relación con las diferentes variables clínicas, los motivos de suspensión de tratamiento y, por último, si la eficacia en la vida real es comparable a la reportada en los ensayos clínicos. Por ello decidimos realizar un estudio observacional retrospectivo en 4 centros que cuentan con equipos multidisciplinarios especializados en enfermedades pulmonares intersticiales (EPI). El objetivo principal de este estudio es conocer el perfil de seguridad de pirfenidona en los pacientes con FPI de nuestra población. Como objetivos secundarios nos proponemos describir los motivos de suspensión del tratamiento y valorar su eficacia.
Materiales y métodosDiseño del estudio y descripción de las variablesEstudio observacional retrospectivo, no patrocinado, realizado en 4 centros de Argentina que cuentan con equipos especializados en EPI. Se incluyeron aquellos pacientes con diagnóstico de FPI según el consenso ATS/ERS/JRS/ALAT 2011, que recibieron pirfenidona en el periodo comprendido entre junio de 2013 y septiembre de 20161. Se excluyeron aquellos pacientes que no completaron al menos 3 meses de tratamiento. Entre las variables clínicas y demográficas se estudiaron la edad, el género, el tabaquismo, la presencia de síntomas de reflujo gastroesofágico (RGE), patrón en la tomografía de tórax de alta resolución (TCAR), datos relacionados con el laboratorio inmunológico, necesidad de biopsia pulmonar para llegar al diagnóstico y desarrollo de exacerbación aguda de la enfermedad. El patrón en TCAR se categorizó según lo propuesto en el consenso ATS/ERS/JRS/ALAT 2011 entre neumonía intersticial usual (NIU) típica, NIU posible e inconsistente con NIU. Se estudió la presencia de enfisema asociado, considerado significativo cuando alcanzó una extensión mayor al 10% en la TCAR. En todos los pacientes se estudiaron el factor reumatoide (FR) (nefelometría, punto de corte 35U/ml) y anticuerpos antinucleares (ANA) (inmunofluorescencia indirecta, punto de corte 1/80). La presencia de exacerbación aguda se definió según criterios propuestos recientemente por un grupo de estudio internacional10. Se estudiaron los siguientes parámetros de función pulmonar y tolerancia al ejercicio: CVF % del predictivo, metros recorridos en el test de marcha de 6minutos, DLCO % del predictivo ajustado a hemoglobina. Todas estas variables se evaluaron al momento del diagnóstico, en la fecha de inicio de la pirfenidona y durante el curso del tratamiento, acorde al criterio de cada grupo médico. Se realizaron las pruebas de acuerdo con lo propuesto por sociedades de referencia11. Con respecto al tratamiento se evaluó el porcentaje de pacientes que recibió inhibidores de la bomba de protones y/o N-acetilcisteína. También se estudió el porcentaje de pacientes que recibió esteroides sistémicos, azatioprina y N-acetilcisteína durante al menos 3 meses12. Para analizar la demora entre el inicio de la enfermedad y el diagnóstico se consideraron los días entre el inicio de los síntomas (disnea y/o tos) y la fecha del diagnóstico en un centro de referencia. Analizamos a su vez el tiempo transcurrido entre el diagnóstico y la fecha de inicio de la pirfenidona. Para el estudio de la tolerancia y seguridad de la pirfenidona se consideraron los datos reportados en las historias clínicas, como efectos adversos (EA), en el periodo de tratamiento comprendido entre la fecha de inicio del fármaco hasta la fecha de la última visita, muerte o suspensión del fármaco. Se definió elevación significativa de las enzimas hepáticas cuando estas se elevaron 3 veces por encima de su valor normal en pacientes que referían síntomas, o 5 veces en aquellos que no referían síntomas. Se compararon las diferentes variables clínicas, funcionales, tomográficas y evolutivas entre los pacientes con y sin EA reportados. Para estudiar la eficacia de la pirfenidona se comparó el cambio en la CVF% en el periodo previo a recibir el fármaco (período pre pirfenidona) con el cambio en la misma variable durante el período en que los pacientes estuvieron expuestos a pirfenidona (período pospirfenidona). Para este análisis se incluyeron solo aquellos pacientes que disponían resultados de CVF% en ambos periodos, con una diferencia no mayor a 6 meses entre los mismos. Finalmente se comparó el cambio en la CVF% durante el tratamiento con pirfenidona entre los subgrupos de pacientes con CVF≥75% y aquellos con CVF<75% en el momento del inicio del tratamiento.
Análisis estadísticoLas variables categóricas se describieron según frecuencia, y las continuas con media y desviación estándar (DE) o mediana y rango intercuartílico (RIC), dependiendo de la presencia o no de distribución normal respectivamente. Para el análisis comparativo se utilizó el test de Chi cuadrado, test exacto de Fisher, el test de Student o Wilcoxon, según correspondiera en función del tipo de variable y su distribución. Se consideró significativa una p<0,05. Se siguió la iniciativa STROBE para comunicar los resultados13.
ResultadosDescripción de la cohorteLas características de la cohorte se muestran en la tabla 1. Se incluyeron 50 pacientes. La distribución de pacientes según el centro fue: 33 pacientes (66%) del Hospital de Rehabilitación Respiratoria María Ferrer de la Ciudad de Buenos Aires, 8 pacientes (16%) del Hospital Privado de Comunidad de Mar del Plata, 5 pacientes (10%) del Hospital de Clínicas José de San Martin de la Ciudad de Buenos Aires y 4 pacientes (8%) del Laboratorio de Función Pulmonar de Alta Complejidad de la Ciudad de Bahía Blanca. La mayoría fueron hombres (76%), con una media (DE) de edad de 67,8 (8,36) años. En 12 casos (24%) fue necesario realizar una biopsia pulmonar quirúrgica para llegar al diagnóstico, de los cuales 8 presentaban un patrón tomográfico de NIU posible, uno de ellos inconsistente con NIU y 3 un patrón de NIU típico. Trece pacientes presentaban enfisema en la TCAR, todos ellos fumadores. De los 37 pacientes restantes que no mostraban enfisema solo 12 (32,4%) nunca habían fumado. En todos los casos el diagnóstico de FPI fue posterior a la discusión de un comité de expertos multidisciplinario y descartando minuciosamente enfermedades del tejido conectivo ocultas y/o neumonitis por hipersensibilidad crónica14. Solo 8 pacientes (16%) presentaban ANA positivo, todos con títulos bajos (menor o igual a 1/160). Catorce pacientes (28%) fueron incluidos en lista tentativa de trasplante pulmonar. Se registraron los parámetros de función pulmonar en 49 pacientes. La media (DE) de CVF% basal fue 69,7 (14,22). En 37 pacientes se midió la DLCO%, con una media (DE) de 44,9 (77,76). En 29 pacientes se obtuvo el valor de PO2, con una media (DE) de 71,6mmHg (25,03). En 42 pacientes se realizó test de marcha de 6minutos, con una media (DE) de metros recorridos de 403,69 (98,33). Fallecieron 13 pacientes (26%) en el periodo analizado, todos por causa relacionada a la FPI. Cinco de ellos (38,46%) fallecieron con posterioridad a una exacerbación y 8 (61,53) por progresión de la enfermedad. Se compararon las características entre aquellos pacientes que fallecieron y los que no. En el grupo de fallecidos 5 pacientes (38,46%) presentaron una exacerbación, mientras que no hubo ningún paciente con exacerbación en el grupo de no fallecidos, alcanzando una diferencia estadísticamente significativa, p=0,0006. Además, 9 pacientes (69,23%) suspendieron el tratamiento transitoriamente en el grupo de fallecidos vs 9 (24,32%) en el grupo de no fallecidos, alcanzando también una diferencia estadísticamente significativa, p=0,005 (tabla 2). Finalmente solo un paciente recibió un trasplante unipulmonar por progresión de la FPI.
Características basales de la cohorte de pacientes con FPI en tratamiento con pirfenidona (n=50)
| Hombres, n (%) | 38 (76) |
| Edad en años, media (DE) | 67,8 (8,36) |
| Biopsia pulmonar, n (%) | 12 (24) |
| Enfisema asociado, n (%) | 13 (26) |
| TCAR típica de NIU, n (%) | 34 (68) |
| TCAR NIU posible, n (%) | 15 (30) |
| TCAR inconsistente con NIU, n (%) | 1 (2) |
| No fumadores, n (%) | 12 (24) |
| Exfumadores, n (%) | 36 (72) |
| Fumadores actuales, n (%) | 2 (4) |
| FR positivo, n (%) | 1 (2) |
| ANA positivo, n (%) | 8 (16) |
| Síntomas de RGE, n (%) | 27 (54) |
| CVF%, media (DE) | 69,79 (14,22) |
| DLCO%, media (DE) | 42,21 (20,37) |
| Metros % recorridos en Test de marcha de 6min, media (DE) | 79,42 (17,57) |
CVF: capacidad vital forzada; DLCO: difusión de monóxido de carbono; FAN: factor anti núcleo; FR: factor reumatoide; NIU: neumonía intersticial usual, RGE: reflujo gastroesofágico; TCAR: tomografía computada de alta resolución.
Comparación entre los pacientes que fallecieron (n=13) y aquellos que no (n=37)
| Pacientes fallecidos (n=13) | Pacientes no fallecidos (n=37) | p | |
|---|---|---|---|
| Edad en años, media (DS) | 67,61±9,43 | 67,91±8,08 | 0,91 |
| Sexo femenino, n (%) | 4 (30,77) | 8 (21,62) | 0,26 |
| Fumadores (actuales o ex), n (%) | 9 (69,23) | 29 (78,28) | 0,58 |
| Síntomas de RGE, n (%) | 3 (23,08) | 24 (64,86) | 0,33 |
| Triple terapia previa, n (%) | 4 (30,77) | 5 (13,51) | 0,16 |
| CVF%, media (DE) | 65,33 (12,97) | 71,24 (14,48) | 0,08 |
| DLCO%, media (DE) | 44,11 (14,60) | 45,15 (18,90) | 0,70 |
| Metros % recorridos en Test de marcha de 6minutos, media (DE) | 83,10 (20,19) | 77,82 (16,54) | 0,32 |
| Tratamiento anti-RGE, n (%) | 11 (84,61) | 35 (94,59) | 0,57 |
| N-acetilcisteína, n (%) | 3 (23,08) | 7 (18,92) | 0,51 |
| Días de exposición a pirfenidona, media (DE) | 1.984,61 (288,23) | 2.092,64 (304,82) | 0,20 |
| Efectos adversos total, n (%) | 5 (38,46) | 10 (27,03) | 0,33 |
| Suspendió tratamiento, n (%) | 9 (69,23) | 9 (24,32) | 0,005 |
| Exacerbación, n (%) | 5 (38,46) | 0 (0) | 0,0006 |
| Enfisema asociado, n (%) | 3 (23,08) | 10 (27,03) | 0,54 |
En negrita: diferencias estadísticamente significativas.
CVF:capacidad vital forzada; DLCO: difusión de monóxido de carbono; RGE: reflujo gastroesofágico.
La media (DE) de días desde el inicio de los síntomas hasta el diagnóstico de FPI fue de 677,7 (563,4), mientras que la media (DE) de días entre el diagnóstico y el inicio del tratamiento con pirfenidona fue de 283,40 (201,7).
Análisis de los efectos adversosLa media (DE) de días de tratamiento con pirfenidona fue 645,68 (428,19), con una dosis media (DE) diaria de 2.064,56mg (301,49). Veintiséis pacientes (52%) recibieron tratamiento con pirfenidona por un período mayor a un año y 14 (28%) por más de 2 años. Se reportaron 19 EA en un total de 15 pacientes (30%). De estos, 11 presentaron un solo evento adverso y 4 pacientes 2 eventos adversos (tabla 3). Al analizar las diferentes variables que podrían influir en el desarrollo de los mismos, no encontramos diferencias estadísticamente significativas entre ambos grupos (tabla 4). Un total de 18 pacientes (36%) interrumpieron el tratamiento, 17 de forma transitoria por menos de un mes, reanudando el tratamiento a dosis plena y uno de ellos definitivamente. Los motivos de suspensión se describen en la figura 1. De estos 18 pacientes solo 3 suspendieron el tratamiento debido a eventos adversos. En 2 pacientes fue suspendido por menos de un mes, siendo en un caso debido a náuseas persistentes, junto a astenia, y en otro por diarrea severa. El tercer paciente suspendió definitivamente por una reacción cutánea fototóxica severa tipo quemadura, que requirió ingreso, presentando posteriormente una evolución favorable. Este último paciente presentaba un fototipo ii de Fitzpatrick (ante la exposición solar se quema con facilidad y se broncea poco), que lo predisponía a dicha fototoxicidad, y no cumplió las medidas de fotoprotección indicadas. Por último, ningún paciente desarrolló hepatotoxicidad.
Efectos adversos reportados en la población de pacientes con FPI bajo tratamiento con pirfenidona
| Efectos adversos total, n (%) | 15 (30) |
| Nauseas, n (%) | 7 (14) |
| Rash cutáneo fototóxico, n (%) | 4 (8) |
| Astenia, n (%) | 5 (10) |
| Diarrea, n (%) | 1 (2) |
| Pérdida de apetito, n (%) | 2 (4) |
| Elevación significativa de la enzimas hepáticasa, n (%) | 0 (0) |
Comparación entre los pacientes que recibieron pirfenidona que presentaron efectos adversos y aquellos que no los presentaron (n=50)
| Con efectos adversos (n=15) | Sin efectos adversos (n=35) | Valor de p | |
|---|---|---|---|
| Edad en años, media (DE) | 67,06 (8,50) | 68,17(8,39) | 0,67 |
| Sexo femenino, n (%) | 5 (33,3) | 7 (20) | 0,25 |
| Fumadores (actuales o ex), n (%) | 10 (66,67) | 28 (80) | 0,71 |
| Enfisema asociado, n (%) | 4 (26,67) | 9 (25,71) | 0,60 |
| CVF%, media (DE) | 63,64 (11,81) | 66,79 (14,15) | 0,43 |
| Síntomas de RGE, n (%) | 6 (40) | 21 (60) | 0,16 |
| Triple terapia previa, n (%) | 3 (20) | 6 (17,14) | 0,54 |
| Tratamiento anti-RGE, n (%) | 15 (32,61) | 31 (67,39) | 0,23 |
| N-acetilcisteína, n (%) | 5 (33,33) | 5 (14,29) | 0,24 |
| Días de exposición a pirfenidona, media (DE) | 609,40 (359,58) | 661,22 (458,44) | 0,67 |
| Dosis final de 2.400mg, n (%) | 5 (22,73) | 17 (77,27) | 0,24 |
CVF: capacidad vital forzada; DLCO: difusión de monóxido de carbono; RGE: reflujo gastroesofágico.

Se incluyeron en este análisis 28 pacientes. No encontramos diferencias significativas entre los periodos prepirfenidona y pospirfenidona. Obtuvimos una mediana (RIC) de seguimiento funcional de 173,5 días en el período prepirfenidona (127,5-265) y de 172 días (144,5-270,5) en el período pospirfenidona (p=0,247). En el análisis comparativo entre ambos periodos (fig. 2) observamos una declinación media (DE) de la CVF% de 4,03% (7,63) durante el periodo prepirfenidona y de 2,64% (7,1) durante el periodo pospirfenidona, sin alcanzar esta diferencia la significación estadística (p=0,534). Esta diferencia tampoco fue estadísticamente significativa en el subgrupo de pacientes con enfisema asociado. Para completar este análisis agrupamos a los pacientes según su severidad y comparamos la declinación de la CVF% durante el tratamiento con pirfenidona (fig. 3). Hallamos 11 pacientes con CVF≥75% (leves) y 17 pacientes con CVF%<75% (moderado y severos). Encontramos una diferencia con tendencia a la significación estadística en el comportamiento de la CVF% a favor del subgrupo con CVF≥75%. Este grupo tuvo un cambio en la mediana (RIC) de CVF% de 1 (–6 a 10), comparado con una mediana (RIC) de –5 (–6 a –1) en el subgrupo <75% (p=0,09). No encontramos diferencias en la proporción de pacientes con enfisema asociado entre ambos subgrupos, siendo 3 (27,2%) en aquellos con CVF≥75% y 3 (17,65%) con CVF<75% (p=0,65).
.")
.")
En este estudio analizamos la seguridad y la eficacia de la pirfenidona como tratamiento de la FPI en la vida real. Observamos eventos adversos en el 30% de la población estudiada. Al igual que en los ensayos clínicos, los mismos fueron leves y con el mismo perfil de eventos gastrointestinales y dermatológicos (14% náuseas y 10% fototoxicidad). La reacción cutánea fototóxica se observó en el 10% de nuestros pacientes, en comparación con un 32% y 28,1% en el estudio ASCEND y CAPACITY, respectivamente3,4. Con respecto a las náuseas, el 14% de nuestros pacientes manifestó haberlas presentado, mientras que la prevalencia fue del 36% en los estudios CAPACITY y ASCEND. Esta diferencia podría explicarse por un menor reporte de efectos adversos por parte de los pacientes en los estudios de vida real, comparado con los ensayos clínicos, presentando en estos últimos una monitorización más exhaustiva. Se podría plantear que la edad avanzada y una menor CVF% al inicio podrían influir en la tasa de eventos adversos y en la tolerancia a la pirfenidona. En este sentido nuestra población no fue diferente a la de los ensayos clínicos antes mencionados. A modo de ejemplo, hallamos en nuestros pacientes una media (DE) de CVF% basal de 69,79 (14,22) y una media de edad (DE) de 67,8 años (8,36), que fue similar a las del estudio ASCEND, que reportó una media (DE) de CVF% de 67,8 (11,2) y de edad de 68,.4 años6,7.
En relación con la supresión por efectos adversos (fig. 1), nuestro estudio estuvo por debajo de lo reportado en otros estudios de la vida real, donde solo 3 pacientes (6%) interrumpieron el tratamiento por eventos adversos. En un estudio español suspendieron por eventos adversos el 14%, mientras que en otro estudio italiano similar este porcentaje alcanzó el 16%15,16. Sin embargo, los ensayos clínicos que respaldaron la aprobación de la pirfenidona para pacientes con FPI reportaron tasas mucho menores de suspensión del fármaco por toxicidad que los estudios de vida real. En el estudio CAPACITY tan solo 3 pacientes (0,9%) debieron suspender la pirfenidona por eventos adversos, siendo todos los casos por reacciones fototóxicas tipo «quemadura»3,4. Este hecho pone en evidencia que, si bien la tasa reportada de EA en los ensayos clínicos es mayor, se trata de eventos en su mayoría leves, que no comprometen la continuidad del tratamiento. Un hallazgo alarmante encontrado en nuestro estudio es que el motivo de suspensión transitorio más frecuente fue la falta de entrega de la pirfenidona por parte de los proveedores (en general seguros médicos). Este es el primer estudio de la vida real que reporta esta problemática reflejando la dificultad que experimentan tanto pacientes como médicos para mantener la continuidad del tratamiento con pirfenidona en la vida real. Estudiamos específicamente la existencia de alguna variable que pudiera relacionarse con el desarrollo de EA (tabla 3). Con este propósito analizamos el comportamiento de variables relacionadas con la severidad de la enfermedad, la utilización de otros tratamientos previos o concomitantes, las dosis y el tiempo de exposición a la pirfenidona, entre otras. No hallamos asociación estadísticamente significativa entre estas variables y el desarrollo de efectos adversos. En este aspecto nuestro estudio se diferencia de otros estudios de la vida real, en los que la adición de otros fármacos se asoció con una mayor tasa de efectos adversos que el tratamiento con pirfenidona como monoterapia8. En relación con la mortalidad, si bien este no es un estudio diseñado para realizar un análisis de tiempo al evento, lo cual nos impide analizar la sobrevida, obtuvimos información de los pacientes fallecidos que nos parece importante reportar. Hallamos en este grupo de pacientes una mayor proporción de exacerbaciones de la enfermedad y de suspensión transitoria del tratamiento antifibrótico. Es importante volver a destacar que el motivo de suspensión transitorio más frecuente fue la falta de entrega de la pirfenidona por parte de los proveedores, lo cual probablemente coloca a los pacientes en una situación de mayor riesgo. En lo que respecta a la eficacia de la pirfenidona, no hemos observado una diferencia significativa en la caída de la CVF% comparando los periodos pre y pospirfenidona (fig. 2). Es interesante que el subgrupo de pacientes con una CVF%≥75 al diagnóstico (leves) presentó una menor declinación de la CVF% con el uso de pirfenidona, mostrando este resultado una tendencia a la significación estadística (fig. 3). En un análisis post hoc de los ensayos clínicos CAPACITY y ASCEND se encontró la misma eficacia en el tratamiento con pirfenidona en el subgrupo de 146 pacientes que presentaba una CVF≥80% con relación a aquellos con valores más bajos3,4. Asimismo, nuestros resultados se asemejan a los reportados por el grupo italiano, que comparó la eficacia de la pirfenidona entre pacientes que presentaban una CVF por encima y por debajo del 75%, encontrando un mayor efecto sobre la declinación de la CVF en el primer grupo7. Teniendo en cuenta que la FPI es una enfermedad progresiva e irreversible, y que la pirfenidona ha demostrado disminuir la declinación de la CVF en el tiempo, su mayor eficacia en pacientes con mejor CVF apoya la importancia de un diagnóstico temprano, y del inicio de fármacos modificadores de la enfermedad en estadios tempranos. Los pacientes que presentaban enfisema en la TCAR de tórax fueron excluidos de los ensayos clínicos pivotales. Por este motivo hay poca información disponible con relación al comportamiento de la pirfenidona en este subgrupo de pacientes. En nuestro estudio el 26% de los pacientes presentaba enfisema en la TCAR de tórax. Los mismos presentaron la misma eficacia y proporción de eventos adversos que aquellos sin enfisema, aunque el número de pacientes analizados no tiene el tamaño suficiente como para tener conclusiones definitivas. Resulta interesante que el tiempo de retraso entre el diagnóstico de FPI y el inicio de pirfenidona en nuestro estudio fue de 283,40 (201,7) días, mientras que en un estudio de vida real italiano fue más del doble (aproximadamente 2 años)7. Es posible que esta diferencia se deba a que todos los centros que participaron en nuestro estudio cuentan con equipos especializados en EPI, lo cual podría agilizar el proceso relacionado con el acceso a la medicación. En contraposición, el tiempo entre el inicio de los síntomas y el diagnóstico de la enfermedad fue de 677,7 (563,4) días. Esto refleja el retraso en la consulta a un centro especializado, lo cual es relevante, ya que se ha demostrado que dicho retraso se relaciona directamente con el aumento de la mortalidad17. Nuestro estudio presenta ciertas limitaciones. Se trata de un estudio observacional retrospectivo por lo que no está exento de un potencial sesgo de selección. La posibilidad de este sesgo se minimizó al incluir de forma consecutiva a todos los pacientes que consultaron a los centros participantes. Por tratarse de un estudio retrospectivo y de vida real, cada uno de los centros participantes utilizó un formulario o historia clínica diferente para la recolección de datos. Esto podría determinar la presencia de un sesgo de información, más específicamente un sesgo de reporte. Con respecto al seguimiento funcional solo pudimos comparar la evolución entre los periodos previo y posterior al inicio de la pirfenidona en 28 de los 50 pacientes. Esto se debe a que los tiempos de seguimiento en ambos períodos eran muy desiguales en los pacientes restantes. Si bien este hecho nos quitó poder para estudiar la eficacia del fármaco, este análisis no era el objetivo primario del estudio. A pesar de estas limitaciones, asumimos que nuestros resultados son válidos debido a que se trata de un estudio multicéntrico y no patrocinado. Las diferencias entre nuestro estudio y otros de la vida real destaca la importancia de realizar estudios de este tipo en distintas poblaciones. Nuestros resultados muestran que existe una buena tolerancia a la pirfenidona en pacientes con FPI, apoyando los resultados observados en los ensayos clínicos y otros estudios observacionales de vida real. Con respecto a la eficacia no hemos observado una diferencia significativa en la declinación de CVF% entre los periodos pre y pospirfenidona.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.







