










Introducción
El déficit de alfa-1-antitripsina (DAAT) es la enfermedad congénita potencialmente mortal más frecuente en la edad adulta. A pesar de ello, continúa siendo una enfermedad infradiagnosticada y cuando se llega al diagnóstico se suele hacer en fases muy avanzadas de la enfermedad pulmonar. Por este motivo es importante llamar la atención de los médicos que atienden pacientes con enfermedad pulmonar obstructiva crónica (EPOC) acerca de las recomendaciones de la OMS y de las sociedades científicas ATS/ERS, que indican de forma categórica que se debe realizar la cuantificación sérica de la alfa-1-antitripsina (AAT) a todos los pacientes con EPOC dentro del esquema diagnóstico habitual de esta enfermedad. También es importante señalar cuándo y cómo deben realizarse otras técnicas diagnósticas de laboratorio, como la determinación del fenotipo o del genotipo. Por último, los detalles referentes a la administración del tratamiento sustitutivo con AAT intravenosa deben actualizarse y ponerse al alcance de los médicos que tratan a estos pacientes. Con estos objetivos, un grupo de trabajo de la SEPAR ha desarrollado esta normativa sobre el diagnóstico y el tratamiento del DAAT. En este documento se detalla la importancia epidemiológica de la enfermedad, con especial referencia a los resultados de estudios realizados en España, las características clínicas de los pacientes con DAAT, las pautas actuales de diagnóstico clínico y de laboratorio y las indicaciones del tratamiento sustitutivo. Esperamos que sirvan de apoyo a los médicos clínicos que atienden a pacientes con EPOC y que puedan solucionar las dudas más frecuentes que se plantean al afrontar la atención a los individuos con sospecha o confirmación de un DAAT.
Alfa-1-antitripsina. Características y consecuencias de su déficit
Características moleculares
La AAT es una glucoproteína de 52 kD, constituida por una cadena de 394 aminoácidos y 3 cadenas laterales de hidratos de carbono. El gen que codifica la AAT se expresa fundamentalmente en los hepatocitos1. La AAT es el inhibidor de proteasas más abundante que hay en el suero humano, donde circula en concentraciones de 120-220 mg/dl determinadas por nefelometría. En condiciones normales, el hígado secreta 34 mg/kg/24 h, pero esta cantidad puede incrementarse 2-5 veces en respuesta a procesos inflamatorios, tumorales o infecciosos. La AAT existente en el suero representa solamente el 40% del total de la proteína y el 60% restante se encuentra en el espacio extracelular impregnando los tejidos corporales.
Además de su capacidad inhibitoria de la tripsina2, la AAT es capaz de inhibir la mayoría de las serinproteasas de los neutrófilos, aunque su diana específica es la elastasa del neutrófilo (EN), que es capaz de digerir la elastina, las membranas basales y otros componentes de la matriz extracelular. Aparte de su capacidad antiproteasa, la AAT neutraliza también las defensinas alfa del neutrófilo, el leucotrieno B-4 (LTB-4) y la interleucina-8 (IL-8), ambos potentes quimioatractantes de neutrófilos al foco inflamatorio3. Por otra parte, modula la adhesión de la EN al receptor de membrana fosfatidilserina del neutrófilo, proceso inicial necesario para la apoptosis, y en este sentido la AAT podría desempeñar un papel importante en la resolución de la inflamación. Además de las propiedades anteriores, la AAT posee 9 radicales de metionina, lo que le confiere una importante capacidad antioxidante. La AAT también es capaz de inhibir o enlentecer la replicación e infectividad de los virus del sida y la de otros virus y bacterias. Todos estos datos, en conjunto, indican que la AAT es una molécula antiinflamatoria natural de amplio espectro, cuya función sería modular las reacciones inflamatorias que se producen continuamente en el organismo humano3,4.
El gen de la AAT se transmite por herencia mendeliana simple de manera autosómica codominante mediante 2 alelos, uno de cada progenitor, que se expresan independientemente en los hijos al 50%1. Este gen se caracteriza por su gran polimorfismo. Mediante isoelectroenfoque (IEF) se han identificado hasta la fecha más de 70 variantes, y su detección aumenta con el avance de las técnicas de identificación. El conjunto de variantes es denominado sistema Pi (protease inhibitor). La mayoría de los variantes carecen de significado clínico, pero hay alrededor de 30 que pueden tener repercusiones patológicas1. Las variantes se clasifican de acuerdo con su velocidad de migración electroforética en un campo magnético con distintos gradientes de pH. Los primeros investigadores denominaron M (inicial de medium) a las de velocidad media, F (inicial de fast) a las de velocidad rápida, y S (inicial de slow) a las de migración lenta. Cuando se descubrieron nuevas variantes, se designó con las letras iniciales del alfabeto a las anódicas y con las finales a las catódicas. El alelo normal, presente en más del 90% de los sujetos normales, se denomina PiM. Los alelos deficientes más frecuentes son PiS (que expresa aproximadamente un 50-60% de AAT) y PiZ (que expresa aproximadamente un 10-20% de AAT)1. Los alelos S y Z codifican proteínas anormales, que polimerizan en el hígado, de modo que el 80-90% de las moléculas de AAT-Z y el 40-50% de las AAT-S son retenidas dentro del hepatocito agrupados en polímeros, normalmente degradados por las proteínas del proteasoma.
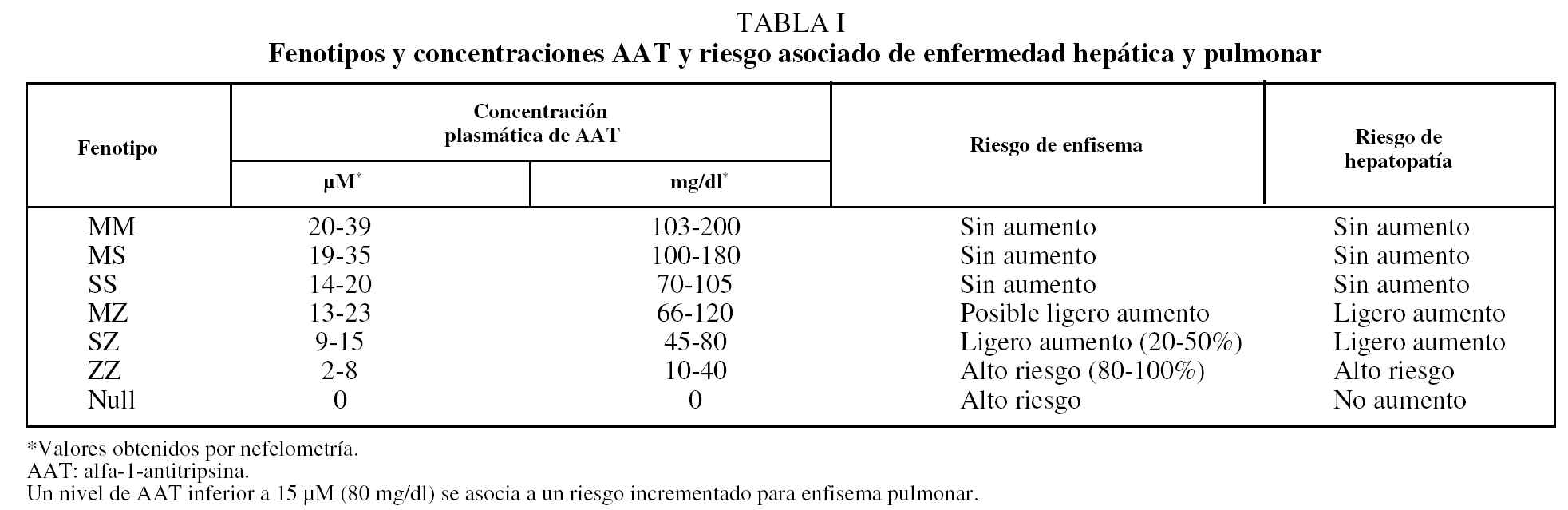
Enfermedades relacionadas con el déficit de la AAT
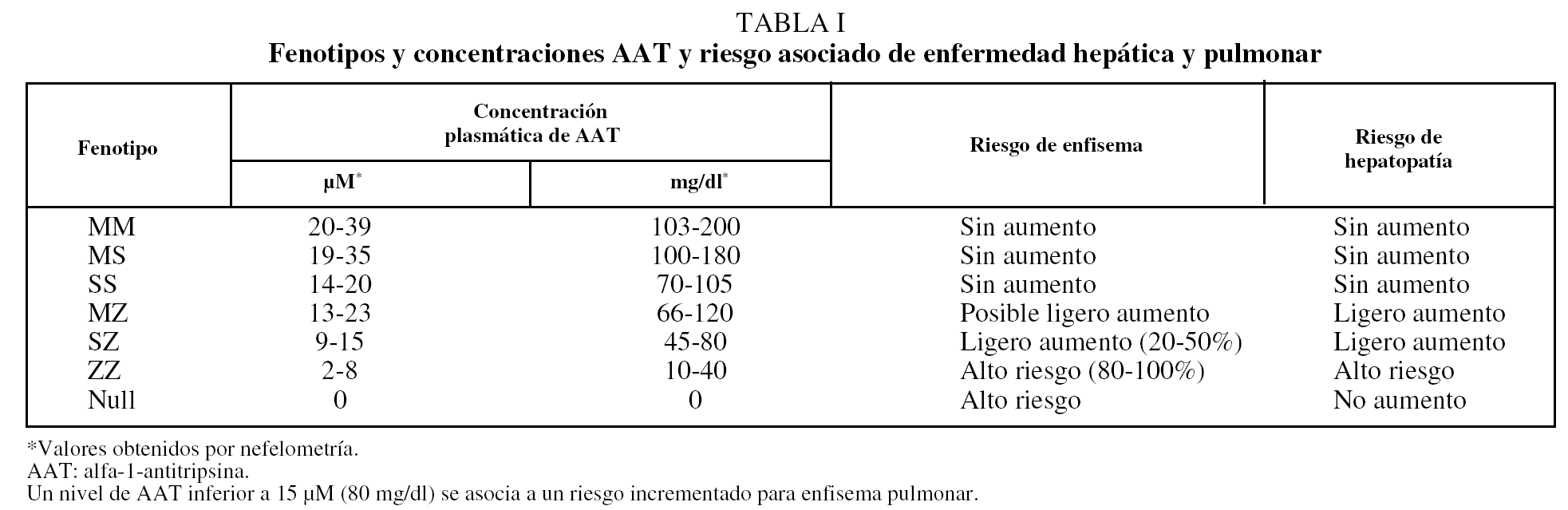
El DAAT confiere una predisposición para desarrollar enfermedades a lo largo de la vida, principalmente enfisema pulmonar y diversos tipos de hepatopatías (incluidas colestasis neonatal, hepatitis juvenil, cirrosis hepática en niños y adultos, y hepatocarcinoma), por lo que se debe considerar como una enfermedad sistémica5,6. Mientras que las hepatopatías se relacionan con la acumulación intrahepática de polímeros, el desarrollo del enfisema es favorecido por las bajas concentraciones plasmáticas y tisulares de AAT, insuficientes para proteger el tejido conectivo del pulmón de los efectos destructivos de las proteasas (tabla I). No existen suficientes pruebas de que la presencia de alelos deficientes influya en la fecuencia o gravedad del asma bronquial7. También se ha sugerido que el DAAT puede estar asociado al riesgo incrementado de desarrollo y progresión de cánceres, incluidos los de vejiga, vesícula biliar, pulmón y linfomas. El mecanismo carcinogénico sería un exceso de proteasas no neutralizado por la AAT, que causaría daño tisular y degradación de la barrera intercelular, hechos que facilitarían el desarrollo y la diseminación del cáncer por la vía del factor de necrosis tumoral.
Existen pruebas de la relación entre DAAT, vasculitis sistémicas y paniculitis necrosante1. El nivel de pruebas disponible sobre la relación entre el DAAT y otras enfermedades como artritis reumatoide, fibromialgia, aneurismas, disecciones arteriales, psoriasis, urticaria crónica, pancreatitis, neoplasias, esclerosis múltiple, etc.1,4 es actualmente bajo, y para obtener conclusiones se necesita realizar más estudios apropiados.
En la práctica clínica, el riesgo de presentar enfermedades se limita a los fenotipos ZZ (96%) y el 4% restante a variantes deficientes raras, denominadas genéricamente M-like y S-like, y a los rarísimos fenotipos nulos1. Los datos existentes sobre penetrancia (porcentaje de individuos con DAAT que presentan enfermedad clínica) son en la actualidad insuficientes, aunque se sabe que prácticamente todos los fenotipos nulos desarrollan enfisema pulmonar, pero no hepatopatía, puesto que al no expresar AAT no generan agregados de polímeros en el hígado8. Hasta un 60% de individuos ZZ puede desarrollar obstrucción crónica al flujo aéreo1 y el factor de riesgo más importante será el grado de tabaquismo, lo que indica que el DAAT, por sí solo, no suele ser suficiente para desarrollar enfermedad y deben existir otros factores genéticos y ambientales favorecedores9.
Epidemiología
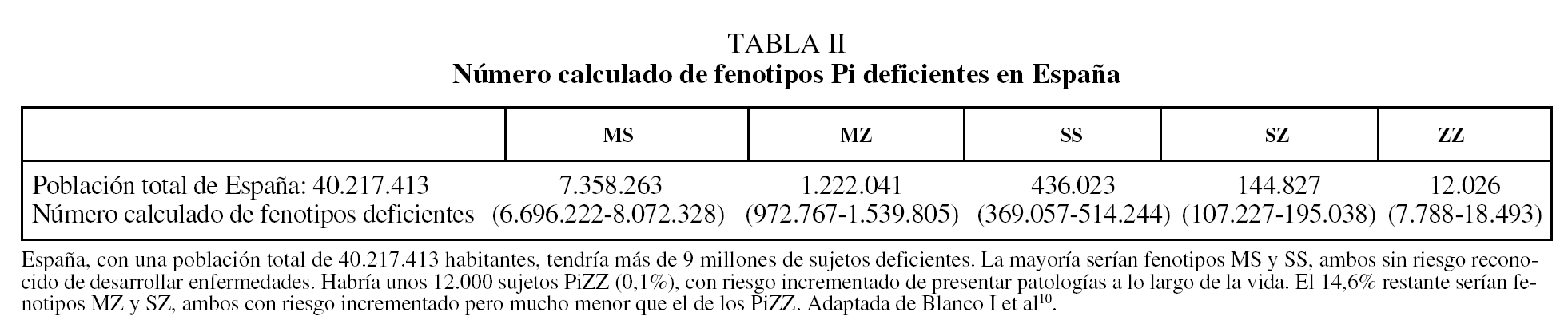
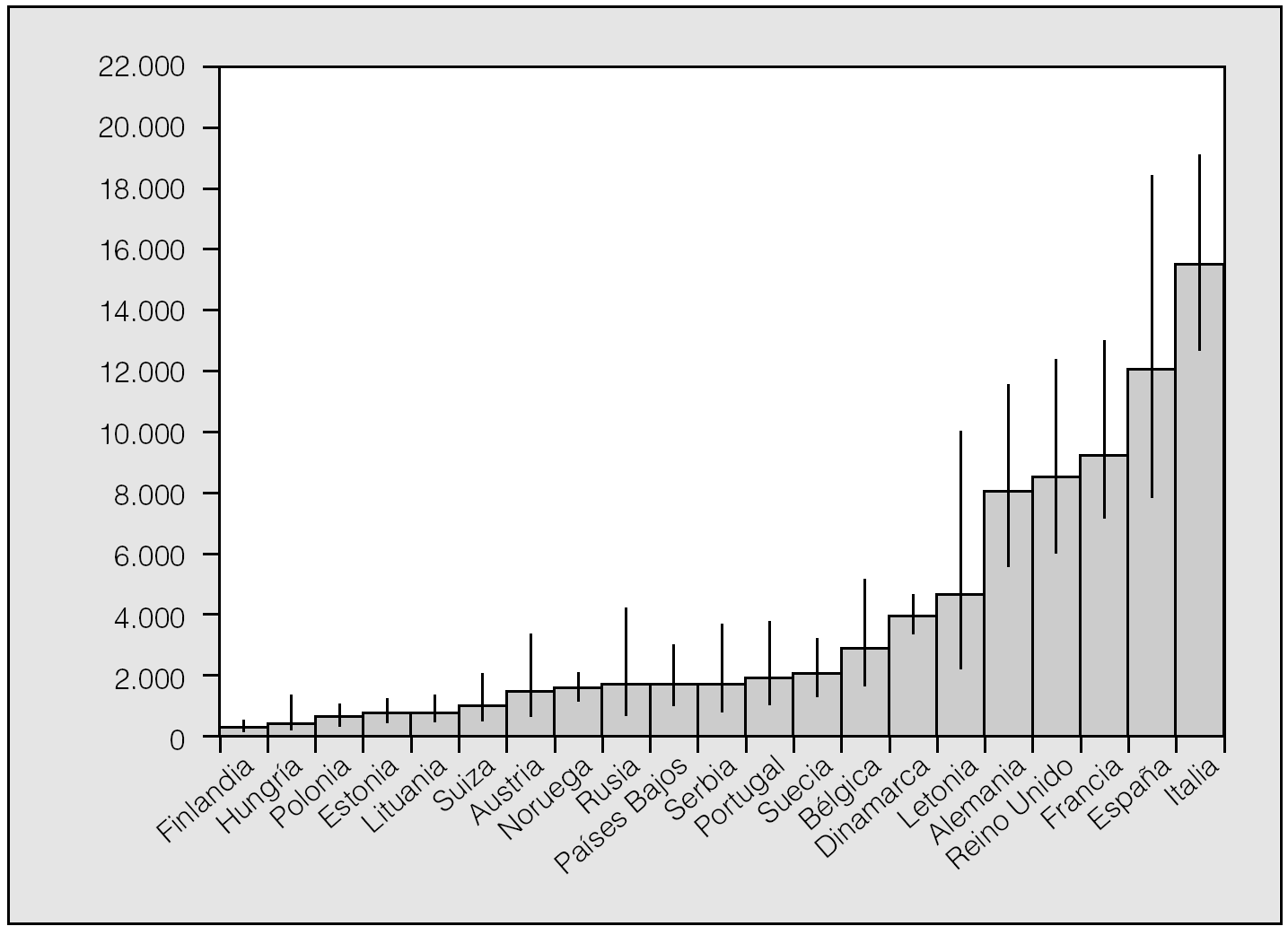
De acuerdo con los resultados de un metaanálisis reciente10, las frecuencias alélicas de S y Z (expresadas en tanto por mil) serían en España: 104 para PiS y 17 para PiZ. Extrapolando estos datos al total de la población española, se calcula que habría en nuestro país un total de 9.173.181 individuos con fenotipos heterocigotos de AAT (intervalo de confianza [IC] del 95%, 9.167.966-9.178.398), con la siguiente distribución: MS, 7.358.263; MZ, 1.222.041; SS, 436.023; SZ, 144.827, y ZZ, 12.026. De acuerdo con estas estimaciones, la prevalencia global de fenotipos heterocigotos sería de uno por cada 4,4 españoles, con la siguiente distribución fenotípica: MS, 1/5; MZ, 1/33; SS, 1/92; SZ, 1/278, y ZZ, 1/3.344 (IC del 95%, 2.175-5.164) (tabla II). En la figura 1 se recogen los números estimados de sujetos ZZ en diversos países europeos para establecer comparaciones. España tendría unas 12.000 personas con fenotipo deficiente ZZ, y es tras Italia el país con mayor número de déficit grave de AAT de Europa11,12.
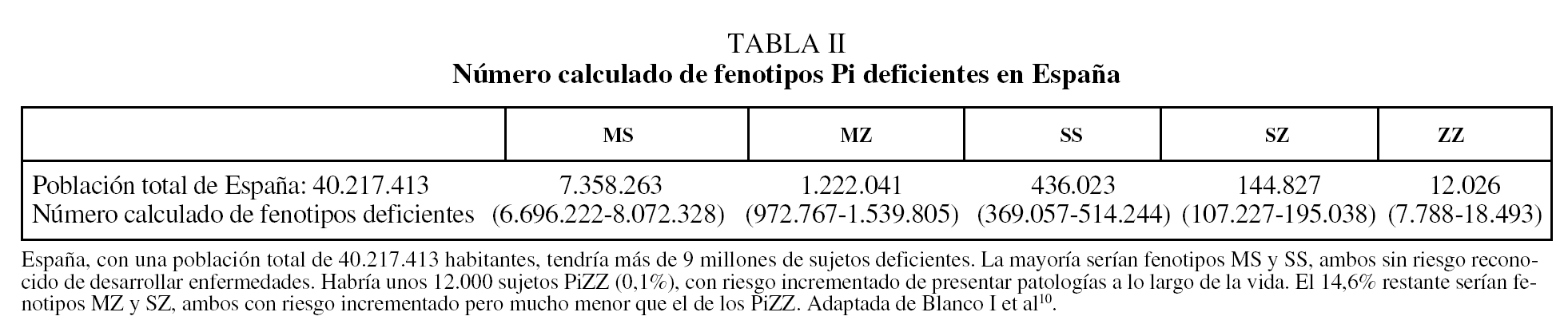
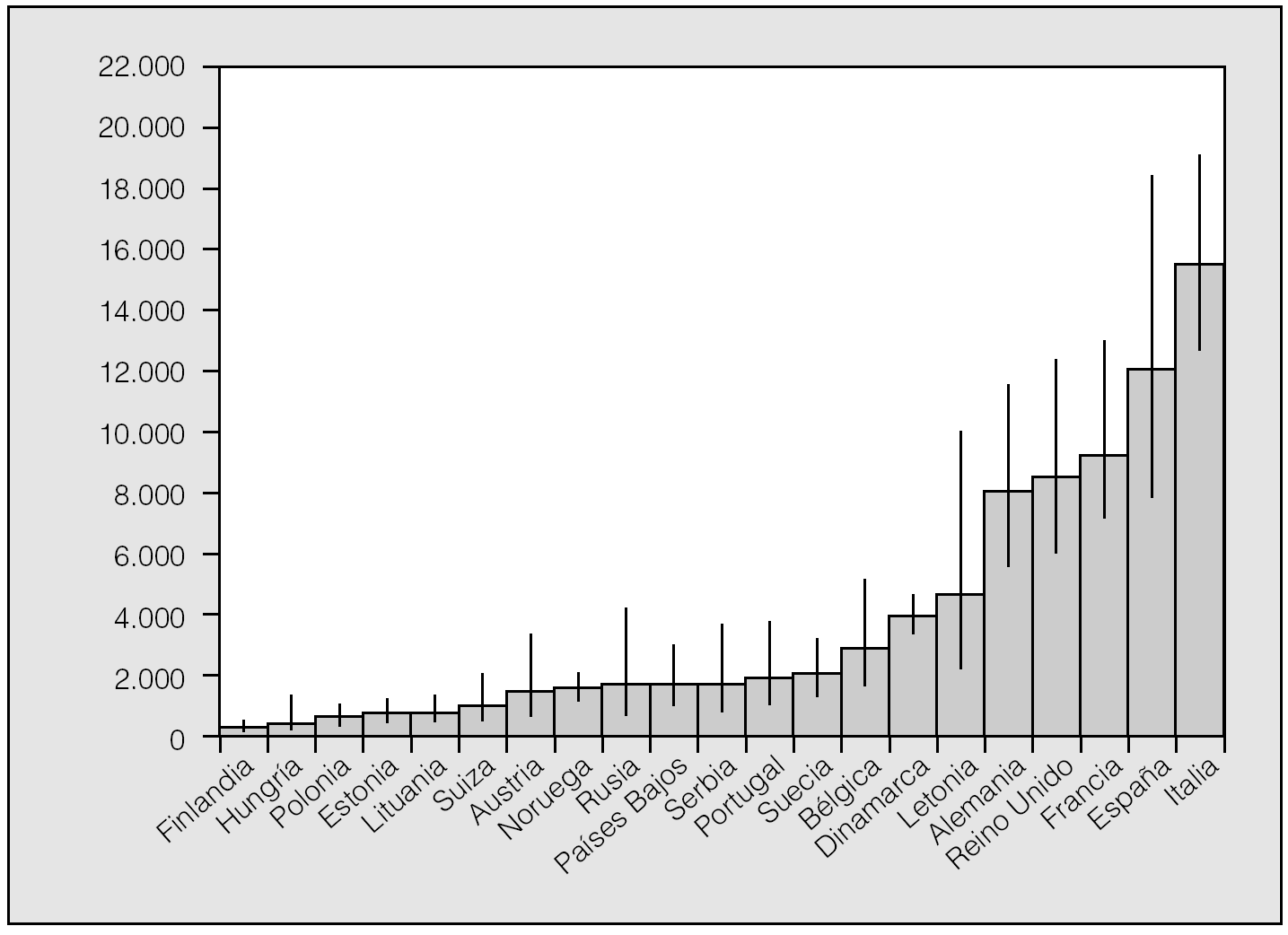
Fig. 1. Número calculado de fenotipos ZZ en 21 países europeos con datos fiables disponibles. Adaptado de Blanco et al11.
Evolución clínica y funcional
Existe una importante variabilidad en la edad de inicio de los síntomas respiratorios, que raramente aparecen antes de los 25 años de edad. Esta variabilidad depende del consumo de tabaco, la presencia de hiperreactividad bronquial y las infecciones respiratorias de repetición. En sujetos fumadores con déficit grave de AAT los síntomas aparecen a los 35-40 años de edad, mientras que en los no fumadores lo hacen una década más tarde13. La disnea es el síntoma más frecuente y está presente en el 80-90% de los individuos; un 65-75% refiere sibilancias, ya sea de forma habitual o en relación con infecciones respiratorias, y hasta un 40% presenta tos, productiva o no, en relación con la existencia de bronquiectasias. En el estudio del Registro NHLBI, el 35% de los individuos refería una historia de asma y más del 50% mostraba una prueba broncodilatadora positiva14. Otros estudios que evalúan la presencia de sibilancias, atopia, aumento de IgE sérica y prueba broncodilata-dora positiva como marcadores de asma encuentran 3 o más de estos marcadores en el 22% de los individuos con déficit de AAT, y sólo en el 5% de aquellos EPOC sin déficit de AAT15,16.
El conocimiento de la historia natural de esta enfermedad ha mejorado gracias al estudio y seguimiento de series de pacientes homocigotos PiZZ, pero aún quedan muchos aspectos por aclarar. Durante los primeros años de vida la única amenaza importante para la salud de estos individuos es la posibilidad de aparición de una hepatopatía. En un estudio realizado en 103 adolescentes PiZZ, diagnosticados en un cribado neonatal, no se encuentran diferencias en las pruebas de función pulmonar frente a un grupo control de la misma edad17.
A partir de la segunda década de la vida su historia natural es menos conocida. Ello se debe, entre otros motivos, a la dificultad para extraer conclusiones de estudios que son diferentes en cuanto a diseño y/o por lo que atañe a las poblaciones seleccionadas. Así, por ejemplo, gran parte de las series está formada por individuos fumadores, ex fumadores y no fumadores y el tabaco puede ocultar los efectos de otros factores de riesgo sobre la función pulmonar. Estudios más recientes realizados en individuos PiZZ no fumadores indican que la exposición a la calefacción doméstica de queroseno, las ocupaciones agrícolas, una historia de exposición profesional a irritantes respiratorios, la presencia de sibilancias y las infecciones respiratorias de repetición y neumonías se asocian a una función pulmonar significativamente más deteriorada18,19.
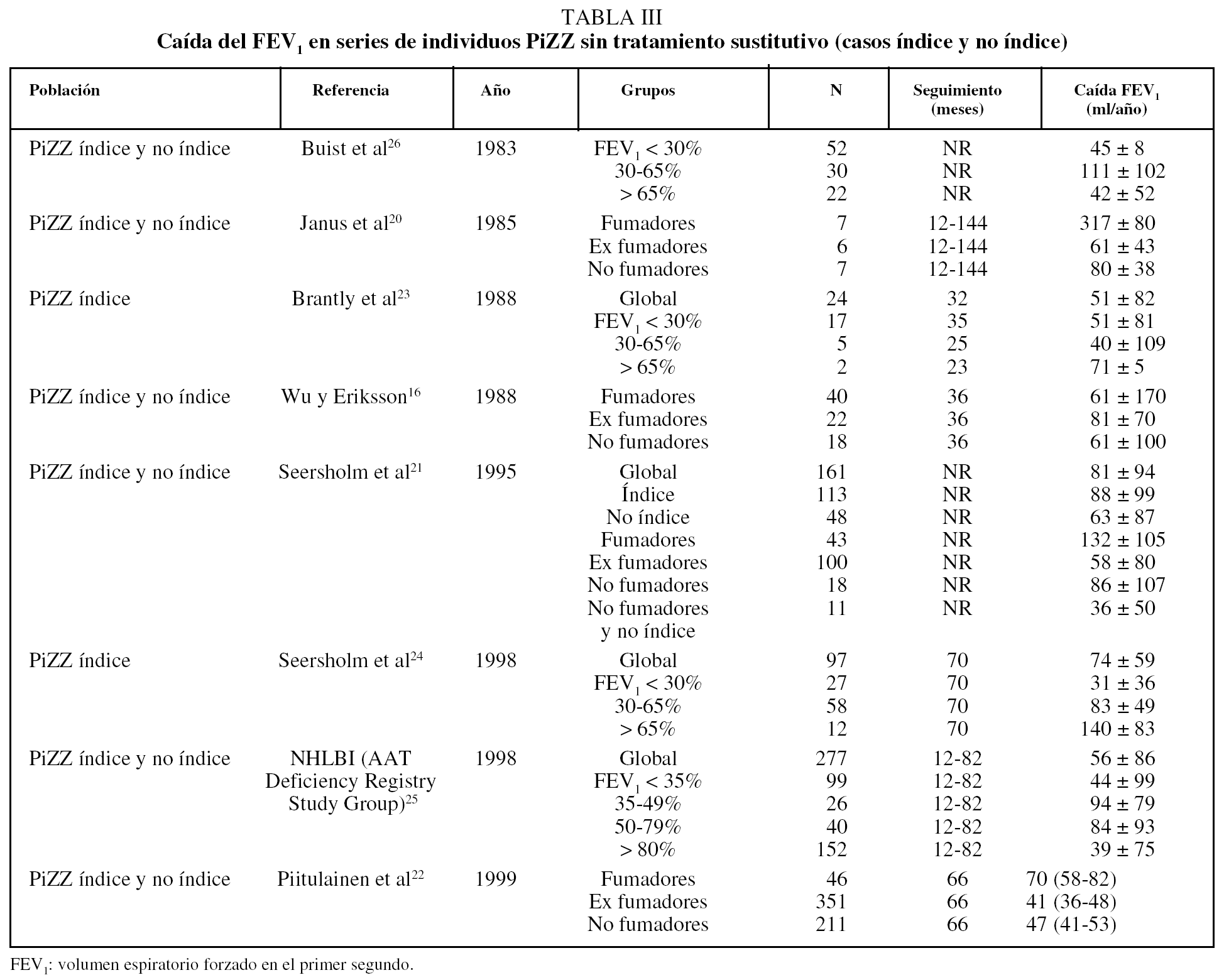
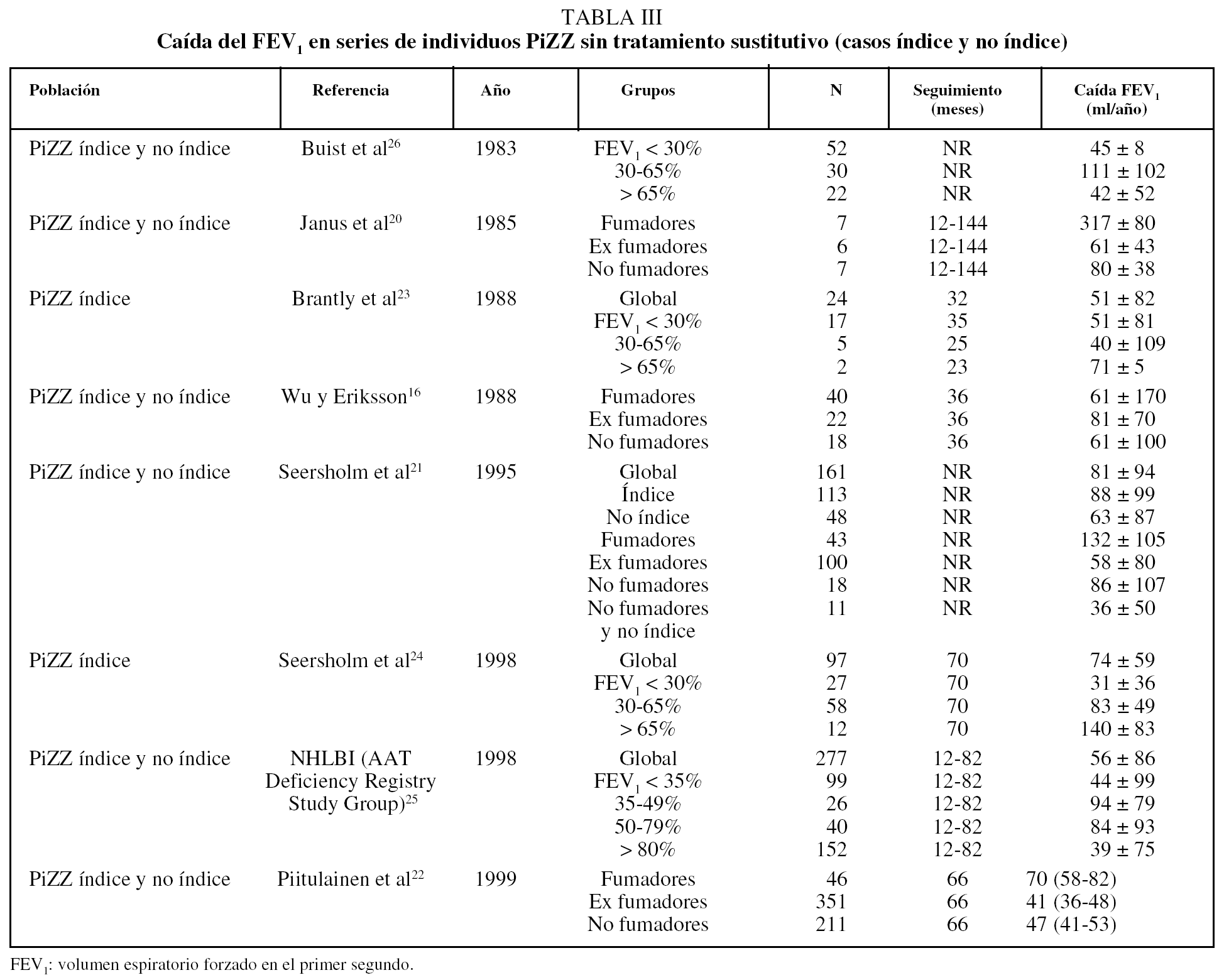
En la tabla III se recoge la caída del volumen espiratorio forzado en el primer segundo (FEV1) en series de individuos PiZZ sin tratamiento sustitutivo (casos índice y no índice). Como se puede observar, en todos estos estudios la caída anual del FEV1 es mayor entre los fumadores y se reduce en los ex fumadores de forma significativa16,20-22; no logra igualar lo que ocurre en individuos con EPOC ex fumadores sin DAAT, en los que la caída del FEV1 alcanza valores similares a los observados en no fumadores (30 ml/año). Los datos son más discordantes en cuanto a los resultados obtenidos según el valor del FEV123-26.
Diferentes estudios han demostrado que el FEV1 es el principal factor pronóstico de supervivencia de los pacientes con déficit grave de AAT16,20,27. La supervivencia a los 2 años es prácticamente del 100% hasta que el FEV1 alcanza el 33% y, a partir de ese momento, desciende de forma exponencial y supone el 50% cuando el FEV1 es el 15% del teórico27. No obstante, un factor que contribuye a resultados confusos y discrepancias entre diferentes estudios es la inclusión de individuos sintomáticos (casos índice) y asintomáticos, que provienen de estudios familiares o programas de cribado (casos no índice). En un estudio realizado en 52 individuos PiZZ, de los que 20 eran asintomáticos, se observó una amplia variabilidad en las pruebas de función pulmonar y se identificaron 3 factores de riesgo de progresión a enfisema: hiperreactividad bronquial, infecciones respiratorias de repetición y factor familiar, pues existía una mayor prevalencia de enfisema entre padres de individuos PiZZ con FEV1 bajo que entre los PiZZ con FEV1 normal13. Estudios recientes indican la existencia de mutaciones genéticas que podrían actuar modificando la historia natural de los sujetos PiZZ. Se ha demostrado un aumento significativo de un polimorfismo en el gen de la sintetasa del óxido nítrico endotelial (C774T) en individuos PiZZ con un FEV1 < 35%28. Rodríguez et al29 encuentran un aumento de la frecuencia de polimorfismos en la glutatión S-transferasa P1 (GST P1-105Val) en pacientes con déficit de AAT que, junto a la edad y el tabaquismo, explicarían el 41% de la variabilidad del FEV1 encontrada en estos pacientes.
La medida de la densidad pulmonar mediante tomografía computarizada (TC) también se ha utilizado como marcador pronóstico en pacientes con déficit de AAT, y su disminución se relaciona con un incremento significativo de la mortalidad a los 5 años30. Otro factor que se ha asociado de forma independiente a la supervivencia en individuos con déficit de AAT es el índice de masa corporal, con un aumento de la mortalidad en individuos con un índice de masa corporal inferior a 2031.
En resumen, el tabaco es el elemento clave en la historia natural de los pacientes con déficit de AAT. El declive del FEV1 y la mortalidad están directamente influidos por la cantidad de tabaco consumido. Los pacientes que dejan de fumar consiguen reducir la caída del FEV1, pero ésta no alcanza las cifras normales detectadas en individuos no deficitarios. Otros factores que condicionan la evolución de la enfermedad pulmonar son la exposición a irritantes profesionales, algunas ocupaciones de riesgo, la presencia de sibilancias, las infecciones respiratorias (en especial las neumonías) y un índice de masa corporal inferior a 20. Existe también un factor de origen genético que condiciona una acumulación de casos índice en determinadas familias. Los individuos PiZZ no fumadores, no casos índice y que no presentan otros factores de riesgo pueden tener una expectativa de vida similar a la de la población general1,32.
Déficit intermedio de la AAT: riesgos de los heterocigotos y otros fenotipos
Los alelos S y Z son los más comúnmente asociados con un déficit intermedio de AAT. Los fenotipos MS y MZ, presentes en las poblaciones caucásicas en un 10% y 3%, respectivamente33, son los que con mayor frecuencia causan un déficit intermedio de AAT y en la actualidad hay una gran controversia sobre si confieren una mayor susceptibilidad que los MM para desarrollar enfisema y EPOC34. Aunque la mayoría de los estudios no han demostrado una mayor prevalencia de síntomas respiratorios o de EPOC en individuos PiMZ no fumadores, 2 estudios realizados en la población general demuestran que dicho fenotipo se asocia con una caída más rápida del FEV1 comparado con individuos MM35,36. Estudios de casos y controles indican un incremento de la prevalencia de individuos PiMZ en los pacientes con EPOC frente al grupo control, y datos del NHLBI Lung Health Study muestran una caída más rápida del FEV1 en fumadores con fenotipo MZ con un riesgo incrementado para EPOC36,37.
El fenotipo SS, que da lugar a concentraciones de AAT del 60% de las variantes normales, no se ha relacionado con EPOC38. Los individuos heterocigotos SZ, que tienen concentraciones de AAT del 40% del valor normal, son poco frecuentes (< 1%) y presentan una mayor prevalencia de EPOC si son fumadores. El grado de afectación funcional sería menor que en los sujetos PiZZ39,40. En la tabla I se muestra el rango de niveles de AAT para cada fenotipo y el riesgo asociado de enfisema.
Diagnóstico clínico
Sospecha diagnóstica
El DAAT puede sospecharse ante diversos cuadros clínicos. Algunos presentan la forma clásica de un adulto joven, fumador o no, con disnea progresiva y enfisema evolucionado; otros se diagnostican en edades más avanzadas tras años de clínica de EPOC con enfisema, y otros están asintomáticos y se diagnostican en los estudios familiares o en cribados epidemiológicos o por la presencia de alteración hepática en la infancia.
Aunque es la enfermedad hereditaria más frecuente diagnosticada en adultos, el hecho de que se inicie de forma tan variada, y que sólo la presente el 1-2% de enfisemas, son algunas de las causas del desconocimiento por parte de muchos médicos, que olvidan solicitar las concentraciones séricas de AAT en muchos enfermos con EPOC o no saben cómo realizar el diagnóstico o dónde o cómo remitirlos para confirmar aquél. Esto provoca en todo el mundo un notable infradiagnóstico de esta alteración genética. En España se calcula un promedio de 10 años entre el diagnóstico de EPOC y el del déficit41 y en Estados Unidos de 7,2 años, y antes de llegar al diagnóstico en un 43% de los casos se había consultado a 3 médicos y en un 12% a más de 642.
Según los datos actuales del Registro Español, en nuestro país se han diagnosticado unos 500 pacientes con fenotipo ZZ, lo que representa un 4% de los 12.000 casos que se calcula que debe haber en España10,43. Estos mismos porcentajes se han diagnosticado en Estados Unidos o en el Reino Unido, y únicamente en Dinamarca se han diagnosticado el 28% de los casos esperados.
La importancia del diagnóstico precoz de esta alteración genética se basa en que permite de forma más precoz hacer un esfuerzo especial en la deshabituación tabáquica, que es determinante en el pronóstico de la enfermedad, tratar los síntomas del enfisema y las agudizaciones, realizar estudios familares para diagnosticar otros casos de forma precoz y dar consejo genético. También se puede iniciar un tratamiento sustitutivo en los casos en que esté indicado.
Clínica y pruebas complementarias
Los enfermos adultos suelen presentarse con los síntomas habituales de la EPOC pero de inicio más precoz: tos, expectoración, disnea y agudizaciones frecuentes, aunque el síntoma capital es la disnea progresiva. Un 60% de los ZZ no fumadores inician los primeros síntomas a los 40 años y un 90% a los 50, aunque los fumadores inician antes la clínica. Por tanto, en muchos casos se hace difícil diferenciarlos de otras causas de EPOC si no se piensa en solicitar la determinación de AAT.
La radiografía y la TC de tórax muestran un enfisema panlobular difuso de predominio en bases. No son habituales las grandes bullas. Una cuarta parte de los casos tiene bronquiectasias asociadas, porcentaje similar al de las series de EPOC44. La función respiratoria puede variar, pero la mayoría tiene un patrón obstructivo con descenso del FEV1 y del cociente FEV1/FVC a unos valores más pronunciados de los esperados por el grado de tabaquismo. También se observa un incremento del volumen residual, hiperinsuflación y descenso de la difusión. Algunos enfermos tienen una prueba broncodilatadora positiva que a veces se asocia con clínica de asma. La hiperreactividad bronquial asociada al fenotipo ZZ ensombrece el pronóstico de estos enfermos.
Durante las agudizaciones se han observado cifras más elevadas de los marcadores inflamatorios IL-8 y del leucotrieno B4 que en las exacerbaciones de la EPOC sin DAAT, lo que podría explicar que las agudizaciones sean frecuentes y pueden ser más graves y prolongadas45.
Algunos individuos homocigotos ZZ inician su sintomatología con una alteración hepática en la primera infancia. Desarrollan una colestasis de gravedad variable, con ictericia y elevación de enzimas hepáticas, y en algunos casos puede evolucionar a una cirrosis con insuficiencia hepática que provoca la muerte si no se intenta un trasplante hepático. Esta afectación hepática se produce por acumulación de la variante Z de la AAT en los hepatocitos, aunque no se sabe por qué sólo una pequeña proporción de homocigotos puede experimentar enfermedad. En la edad adulta la mayoría de los enfermos presenta una función hepática normal.
Otra forma clínica más infrecuente es la paniculitis con nódulos subcutáneos eritematosos y dolorosos generalizados y que pueden ulcerarse.
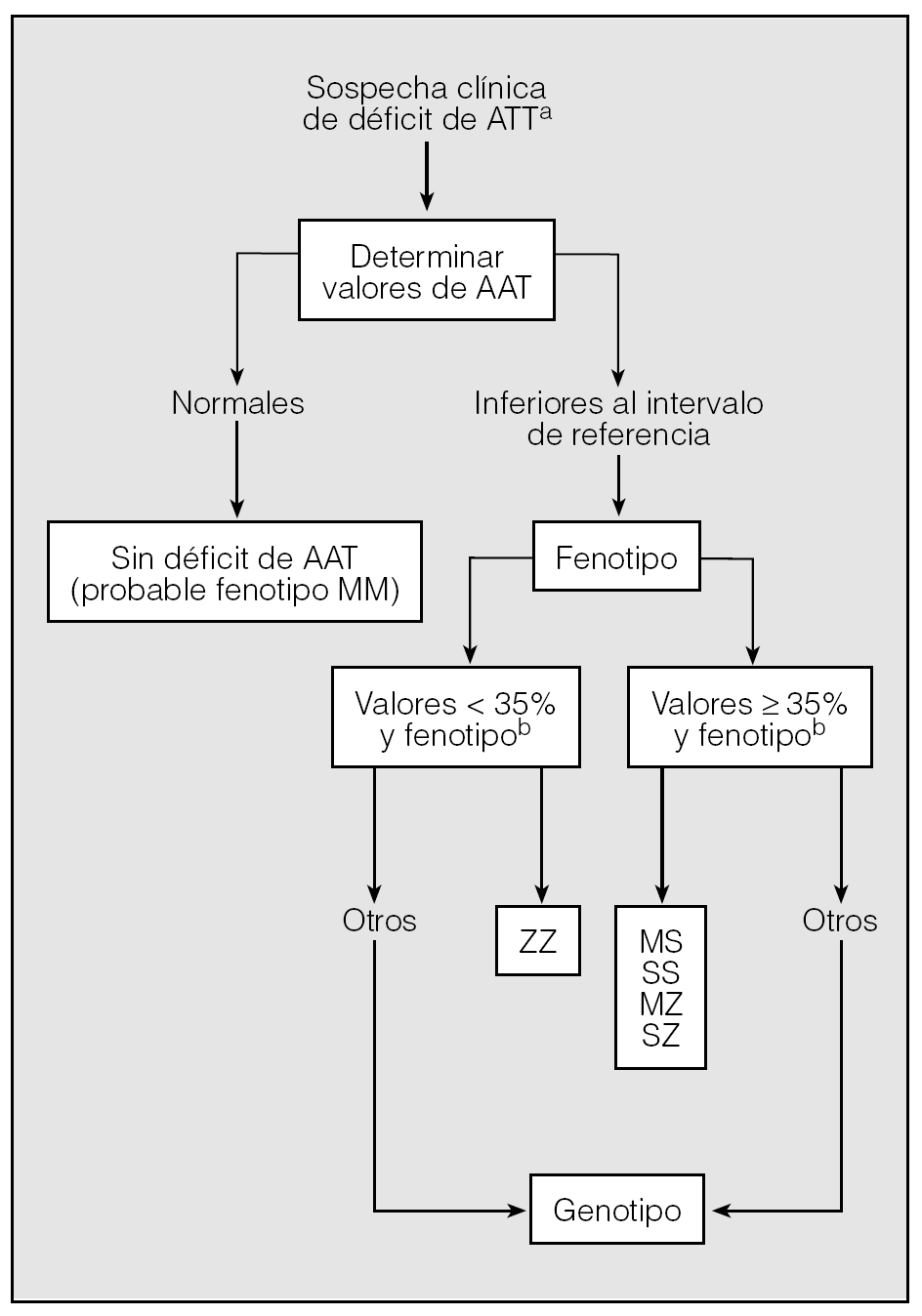
Para llegar a un diagnóstico de seguridad en los casos sospechosos, se pueden seguir los pasos de un algoritmo (fig. 2).
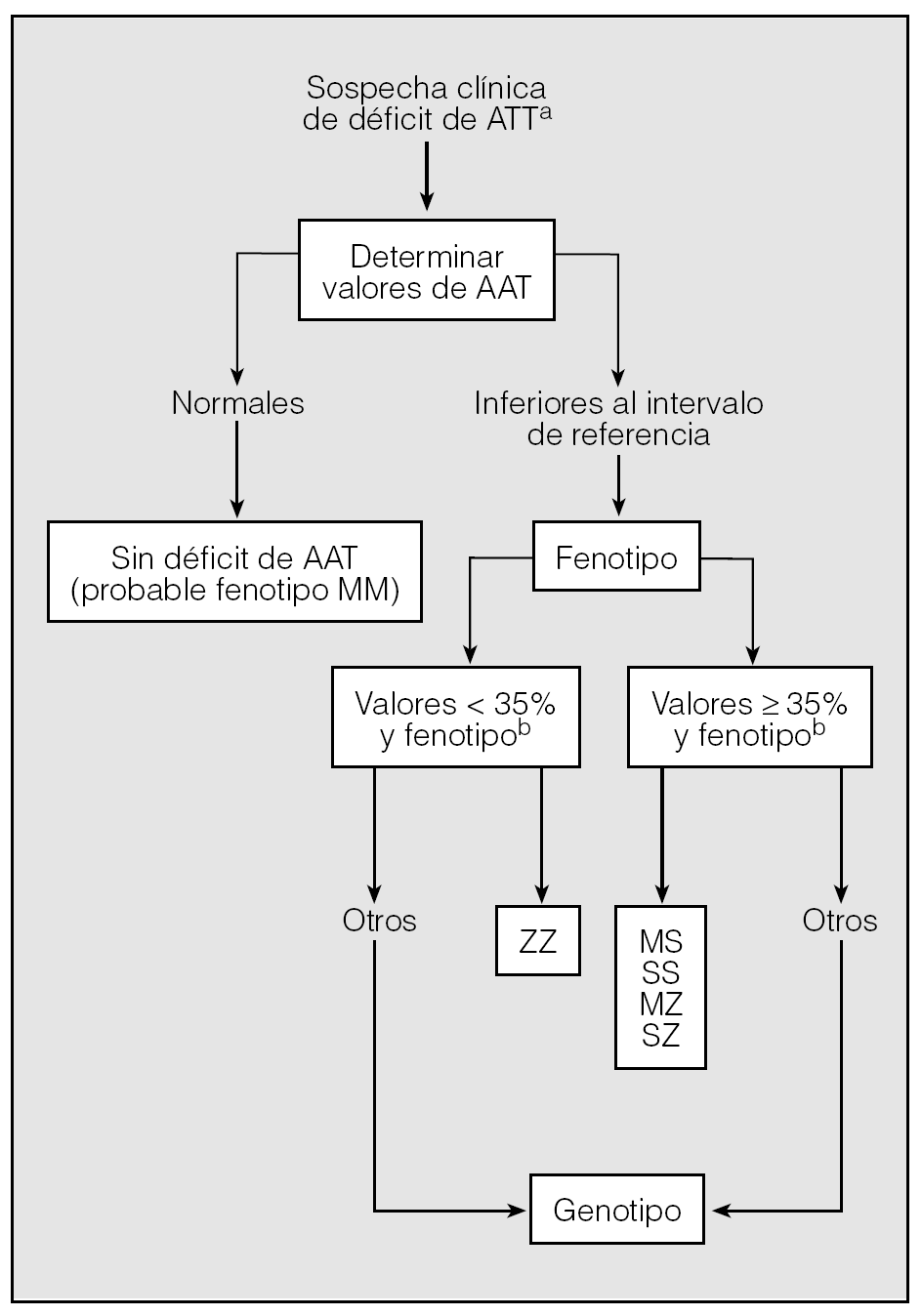
Fig. 2. Algoritmo diagnóstico del déficit de alfa-1-antitripsina (AAT).aVéase tabla IV.bPorcentaje respecto al límite inferior del intervalo de referencia.
Estudios genéticos familiares y cribados poblacionales
La determinación genética en personas con clínica que hace sospechar el DAAT debe realizarse siempre. Las personas no fumadoras ni asmáticas, que presentan disnea y alteración de las pruebas de función respiratoria y los fumadores con alteración de la función pulmonar antes de los 40 años serían los principales candidatos a DAAT. Sin embargo, con estos únicos criterios no se diagnosticarían muchos otros enfermos con una presentación más atípica. Por este motivo, en un consenso patrocinado por la OMS en 1997, se recomendó que a todos los pacientes con EPOC se les realizara, por lo menos una vez en la vida, una determinacion de las concentraciones de AAT, lo que también se incluye en las recomendaciones de la ATS y el ERS1,46. Por tanto, siempre estaría indicada la determinación una vez en la vida de las concentraciones séricas de AAT a diversos grupos de personas que presenten, además de EPOC, alguna de las enfermedades que se recogen en la tabla IV.

La determinación del fenotipo estaría indicada en las personas con valores bajos de AAT y la secuenciación genotípica sólo debería hacerse cuando exite discordancia entre la concentración de AAT y el fenotipo (tabla V).

Antes de la realización de estudios genéticos poblacionales hay que valorar los beneficios: diagnóstico precoz, medidas preventivas, consejo genético y tratamientos específicos; y los potenciales inconvenientes: psicológicos, sociales o laborales para los enfermos y el coste económico y el rigor científico del estudio.
Existirían 2 motivos para determinar las concentraciones séricas de AAT y el fenotipo en grupos de población sana:
1. Estudio en personas predispuestas. Serían personas sin síntomas específicos pero con alta probabilidad de presentar la alteración genética. Se trata principamente de los familiares consanguíneos. El riesgo de ser homocigoto PiZZ en los hijos de 2 heterocigotos con un alelo Z es del 25%, y los hijos de un homocigoto ZZ y un heterocigoto serán todos PiZZ o portadores de un alelo Z. Por tanto, no hay que esperar a establecer el diagnóstico cuando aparezcan síntomas hepáticos o de enfisema, y siempre debe realizarse el estudio de los familiares consanguíneos de los enfermos homocigotos ZZ, de los heterocigotos MZ y SZ o de los portadores de alelos raros deficientes47.
2. Cribados de población general. Son estudios para determinar la prevalencia del déficit en poblaciones diversas. El cribado de recién nacidos o de la población general sólo debería hacerse para conocer la prevalencia poblacional o geográfica cuando se supone que es alta, hay un importante infradiagnóstico y una elevada prevalencia de fumadores.
Sólo deben realizarse a través de registros o sociedades científicas, y requieren un diseño supervisado por expertos y una adecuada información a los participantes, tanto antes como después de conocer los resultados1,48.
Se determinarán las concentraciones de AAT y, si son inferiores a unos valores predeterminados, se investigará el fenotipo.
En todos estos cribados familiares o de poblaciones, cuando se diagnostique un caso deficitario con fenotipo concordante, deberá analizarse con radiografía de tórax, estudio funcional respiratorio y analítica hepática. Se completará con el estudio de sus consanguíneos. Todos los casos identificados de incluirán en el Registro Nacional.
Siempre hay que tener en cuenta la gran diferencia que existe, tanto en la expresión clínica como en el pronóstico, entre los casos-índice, que son los enfermos que se diagnostican por sospecha clínica, y los casos que se descubren en los estudios familiares o los cribados de población general que suelen estar asintomáticos y tienen un mejor pronóstico a largo plazo, sobre todo si siguen las medidas higiénicas preventivas49.
Diagnóstico de laboratorio
El diagnóstico de laboratorio de déficit se realiza en suero mediante la determinación cuantitativa de AAT y la identificación del fenotipo de AAT. El análisis molecular del gen de la AAT o genotipo es el método de referencia para la identificación de las variantes alélicas poco frecuentes. Esta última determinación se realiza en sangre total utilizando EDTA como anticoagulante.
Determinación de la concentración sérica de la AAT
La inmunonefelometría cinética es el método más comúnmente utilizado para la determinación de los valores séricos de AAT. Esta técnica de cuantificación se basa en la formación de inmunocomplejos insolubles entre la proteína y los anticuerpos anti-AAT. Si durante este proceso se hace incidir un rayo de luz intenso, éste es dispersado por las partículas que precipitan y la intensidad de la radiación dispersada es proporcional a la cantidad de antígeno presente en la muestra de suero. El análisis debe de efectuarse, en lo posible, en muestras de suero frescas o congeladas a -20 ºC, evitando congelaciones y descongelaciones repetidas (tabla VI).

Para una buena interpretación de la determinación, cada laboratorio debe haber determinado previamente los valores normales de AAT en muestras de suero obtenidas de población sana. Los valores de referencia de la concentración de AAT para muestras de suero de personas adultas sanas son de 103-200 mg/dl50. Las cifras de AAT en la población infantil son inferiores a las de la población adulta. Los resultados de la determinación cuantitativa pueden también expresarse en unidades micromolares, la conversión a µM se realiza multiplicando la concentración en mg/dl por 0,1923.
La determinación sérica de la AAT constituye la base del diagnóstico del déficit congénito. Valores por debajo del 35% de la normalidad indican posibilidad de estado homocigoto PiZZ.
Para la interpretación del resultado de una determinación cualitativa aislada, hay que tener en cuenta que al ser la AAT un reactante de fase aguda, los procesos infecciosos o inflamatorios pueden alterar la determinación, de manera que se pueden detectar valores normales o altos en pacientes con déficit moderado. También se han descrito valores elevados de AAT durante el embarazo y después del consumo de anticonceptivos orales. Por eso el diagnóstico del DAAT se debe basar en los resultados de la determinación de la concentración de AAT y de la identificación del fenotipo.
Determinación de los fenotipos de la AAT
El gen de la AAT presenta un gran polimorfismo, lo que explica el elevado número de variantes proteicas o fenotipos. Estas variantes se deben en su mayoría a la mutación de una base en la cadena de ADN, lo que se traduce en la sustitución de un aminoácido por otro y en la modificación de la carga proteica.
El método más ampliamente utilizado para la identificación de las variantes proteicas es el IEF. Esta técnica consiste en la separación electroforética de las proteínas, de acuerdo con su punto isoeléctrico, en un gel de acrilamida/bisacrilamida y en un gradiente de pH de 4,2-4,5. Siempre que sea posible, el análisis debe efectuarse en muestras de suero frescas o congeladas a -20 ºC, evitando congelaciones y descongelaciones repetidas. Las condiciones de almacenamiento son muy importantes, ya que influyen en el resultado, en particular para la identificación de la AAT de fenotipo PiZ, los antígenos del cual son muy lábiles y puede degradarse.
La determinación del fenotipo es el método requerido para confirmar el DAAT. Está indicada en los casos con valores de AAT inferiores al rango de normalidad o en los que presenten valores cercanos al límite inferior de este intervalo; estos últimos casos podrían corresponder a un fenotipo MS, SS o MZ. Al igual que se ha comentado para los valores de referencia de AAT, es preciso establecer los valores propios de referencia de la concentración de AAT de cada uno de los fenotipos mayoritarios, as&iac ute; como conocer los fenotipos prevalentes en el área geográfica donde se realizan los estudios. En la tabla I se presentan los resultados de la relación entre el fenotipo y la concentración de AAT. Se observa que los individuos con fenotipo PiMM y PiMS son los que presentan las concentraciones séricas más elevadas de AAT y no hay solapamiento entre la concentración de AAT de los fenotipos PiMM o PiMS o PiSS (poco deficitarios) con la de los fenotipos PiSZ o PiZZ. Únicamente existe solapamiento entre la concentración de AAT de los fenotipos deficitarios PiSZ y PiZZ.
Determinación del genotipo de la AAT
El análisis molecular del gen de la AAT es el método de referencia para identificar las variantes alélicas poco frecuentes relacionadas con el déficit congénito de AAT o para la caracterización de variantes nuevas51-53. Asimismo, es el método más apropiado para identificar las variantes nulas. La determinación se aplica también para el estudio de los casos en los que no existe concordancia entre la concentración de AAT y el fenotipo; sería el caso, por ejemplo, de un paciente con fenotipo normal PiMM y que, sin embargo, presentase un déficit de AAT. Probablemente, se trataría de una variante proteica de AAT con un punto isoléctrico similar al de la variante PiMM, que por esta razón no se podría catalogar mediante la técnica de fenotipificación.
La determinación se lleva a cabo a partir de sangre total, utilizando etilendiaminotetraacetato (EDTA) como anticoagulante. Las muestras pueden conservarse a 4 ºC durante 48 horas después de realizada la extracción, o almacenarse a -20 ºC en caso de que no se procesen inmediatamente (tabla VII). El método más utilizado consiste en realizar una extracción de ADN a partir de las células mononucleares, seguida de su amplificación mediante la técnica de la reacción en cadena de la polimerasa (PCR) y de la secuenciación cíclica de los productos de la PCR. Para esta determinación, es preciso realizar un estudio completo de las secuencias de ADN de los 4 exones del gen que codifica la AAT, así como de las secuencias de los intrones correspondientes54.

Otras determinaciones
Determinación de la capacidad de la AAT para inhibir la elastasa de los neutrófilos
La acción inhibitoria antielastasa está en relación con la concentración de la AAT, de manera que a mayor cantidad de AAT circulante más efecto inhibitorio. Sin embargo, algunos pacientes pueden presentar valores normales o elevados de AAT y poca actividad antielastasa debido a que no toda la proteína es activa. Estos casos están asociados a la AAT oxidada, degradada o con poca capacidad de unión a los neutrófilos.
La determinación de la actividad está indicada principalmente en los siguientes casos: en el estudio de los pacientes con EPOC en los que se sospeche DAAT a pesar de detectarse un valor normal de AAT, para conocer si en la fase de agudización de la EPOC el incremento de AAT es proporcional a la actividad antielastasa, y en general para valorar la implicación del DAAT en la gravedad de la enfermedad pulmonar, ya que muchas veces la concentración no guarda relación con la evolución clínica.
El método para determinar la capacidad inhibitoria antielastasa consiste en incubar muestras de suero con un exceso de elastasa y en valorar la elastasa residual. La actividad antielastasa será mayor en los casos en que se detecte una concentración baja de elastasa residual.
Evaluación del funcionalismo hepático
La evaluación del funcionalismo hepático en los casos con DAAT se realiza mediante las determinaciones de ALT, AST, bilirrubina, albúmina y pruebas de coagulación.
Diagnóstico de déficit mediante el análisis del proteinograma sérico
El diagnóstico de déficit mediante la observación de la reducción o ausencia de la banda de las globulinas alfa-1 en el proteinograma sérico está en desuso; se trata de un método poco sensible y específico, y que requiere siempre la confirmación del resultado mediante los análisis cuantitativo y cualitativo de la AAT55.
Cribado del déficit de la AAT en muestras de sangre seca en papel
Las muestras de sangre seca en papel son especialmente útiles para el cribado y diagnóstico de las enfermedades genéticas. La obtención de las muestras es mínimamente invasiva y son fáciles de almacenar y enviar al laboratorio de referencia donde están centralizadas las determinaciones.
La determinación se realiza a partir de sangre capilar, por punción estéril del pulpejo del dedo. Las gotas de sangre se aplican sobre discos de papel que se dejan secar a temperatura ambiente antes de enviarse por correo al laboratorio central del estudio56 (tabla VIII).

En los protocolos establecidos se determina habitualmente la concentración de AAT mediante nefelometría cinética57 y los genotipos deficitarios más frecuentes S y Z mediante PCR a tiempo real54. Hay que tener en cuenta que la determinación en sangre seca es un método de cribado y, como tal, todos los casos diagnosticados de déficit deben estudiarse en muestras de suero, realizando las determinaciones de AAT y fenotipo48.
Importancia de los registros: Registro Español y AAT Deficiency Internacional Registry
Los registros de pacientes fueron creados por la necesidad de recoger información de grupos amplios de pacientes dada la escasa prevalencia del DAAT. El Registro Danés se fundó en 1978 y en 1994 incluía más de 500 individuos. En Suecia, país pionero en el estudio de esta enfermedad, el registro se fundó en 1991 y en diciembre de 1994 incluía a 665 personas. Sin embargo, el mayor interés por los registros se inició con la disponibilidad del tratamiento sustitutivo, ya que muy pronto se llegó a la conclusión de que no se podría realizar un ensayo clínico sobre la eficacia a largo plazo del tratamiento. En este contexto, algunos registros aparecieron como una alternativa a este ensayo al comparar la evolución de amplias cohortes con y sin tratamiento sustitutivo. Los principales registros creados con esta finalidad fueron el alemán, que se inició en 1989 y que en 1995 recogía información de 443 pacientes de 25 centros, y el registro del NHLBI, que se inició en 1988, incluyendo hasta octubre de 1992 a 1.129 pacientes procedentes de 37 centros de Estados Unidos y Canadá25.
El Registro Español se fundó en 199358 y, dado el pequeño número de pacientes que se esperaba reclutar, sus propósitos iniciales fueron: a) conocer las características y la frecuencia del déficit de AAT en España; b) establecer normativas adaptadas a nuestro país sobre el tratamiento y el seguimiento de pacientes con el déficit; c) ofrecer información a los médicos que tratan a estos pacientes en toda España; d) incrementar el conocimiento y el interés por esta "no tan rara" enfermedad e intentar disminuir el infradiagnóstico o el retraso en el conocimiento del déficit, y e) ofrecer soporte técnico para la determinación del fenotipo Pi y, si es necesario, del genotipo en los individuos con sospecha de déficit de AAT.
En 1997 se constituyó el AAT Deficiency Internacional Registry (AIR) como una iniciativa europea auspiciada por la European Respiratory Society (ERS), pero de alcance más amplio, ya que junto a países europeos como el Reino Unido, Suecia, Dinamarca, Países Bajos, España, Italia, Suiza y Alemania, incluye también representantes de Nueva Zelanda, Australia, Sudáfrica, Argentina, Brasil y Canadá59. Es posible que este registro sea la plataforma de amplias iniciativas destinadas a conocer mejor la historia natural de la enfermedad y, tal vez, el estímulo para realizar ensayos clínicos definitivos sobre la eficacia del tratamiento sustitutivo. Sin duda, iniciativas como los registros nacionales e internacionales son la única posibilidad de reclutar un número suficiente de pacientes para avanzar en el conocimiento del déficit de AAT60.
El registro de los pacientes deficitarios (PiZZ, variantes deficitarias raras y PiSZ) se realizará a través de la web de SEPAR (www.separ.es/air). El Registro Español de pacientes con déficit de AAT se encuentra dentro del área Insuficiencia Respiratoria y Trastornos del Sueño (IRTS) y se puede acceder desplegando la pestaña de "Áreas de Trabajo". En la página web del Registro Español de déficit de AAT podemos encontrar información general del Registro, de los coordinadores del Registro y responsables de diferentes comunidades autónomas, acceso al laboratorio central del Registro y a publicaciones. También existen links con otras webs médicas y de asociaciones de enfermos. El registro de pacientes se realizará mediante acceso personal. Para ello solicitaremos claves y en unos días recibiremos "usuario" y "clave" para nuestro acceso personal (tabla IX y fig. 3).


Fig. 3. Página inicial del Registro Español de pacientes con déficit de alfa-1-antitripsina. Disponible en: http://db.separ.es/cgi-bin/wdbcgi. exe/separ/ air.baseline_pack_3.indice? p_origen=34
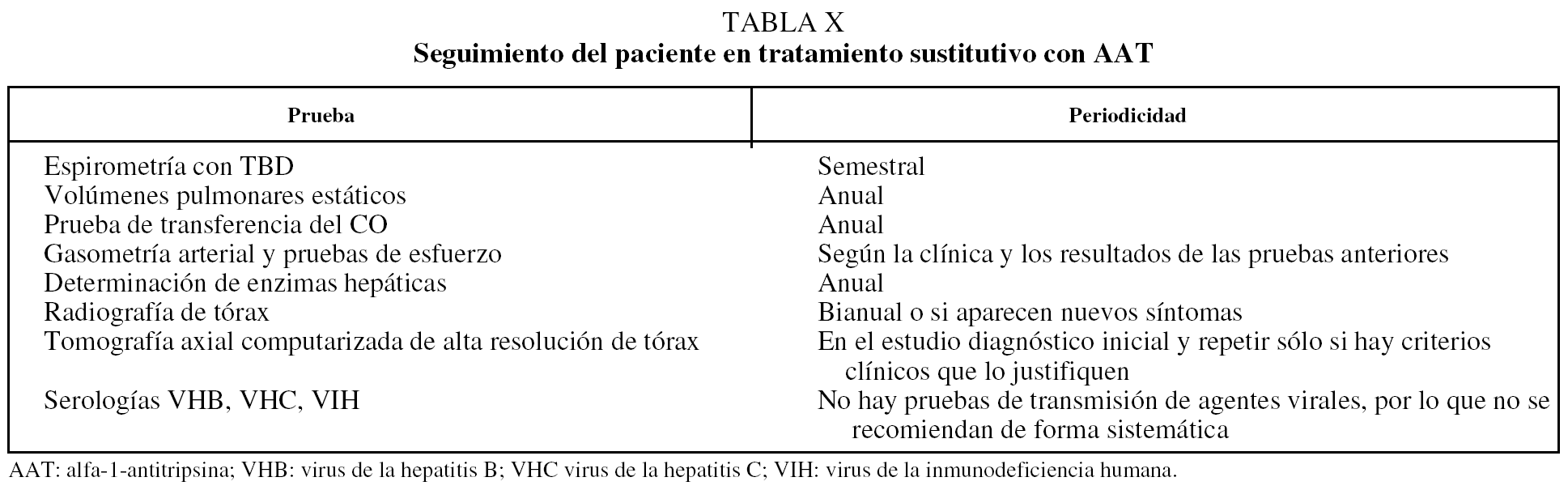
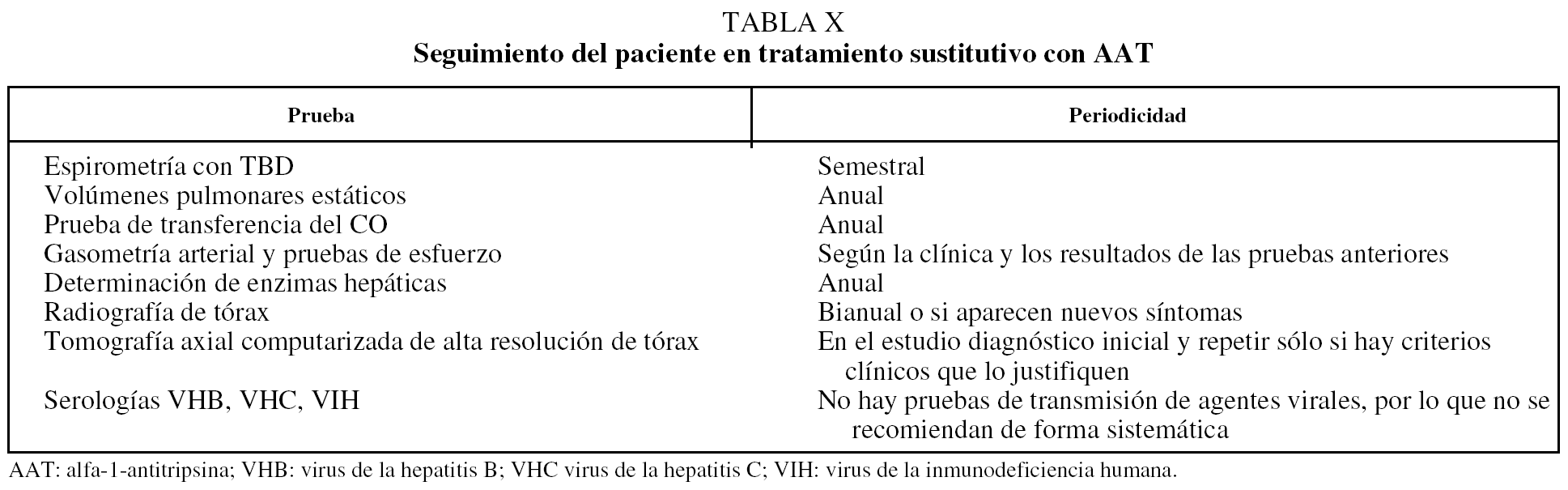
En la tabla X se recogen las pruebas que deberemos realizar de forma periódica. Cada 6 meses actualizaremos los datos de nuestros pacientes en la web del Registro rellenando las hojas electrónicas de seguimiento para lo que utilizaremos nuestro acceso específico.
Tratamiento sustitutivo
Fundamentos del tratamiento sustitutivo. Eficacia bioquímica
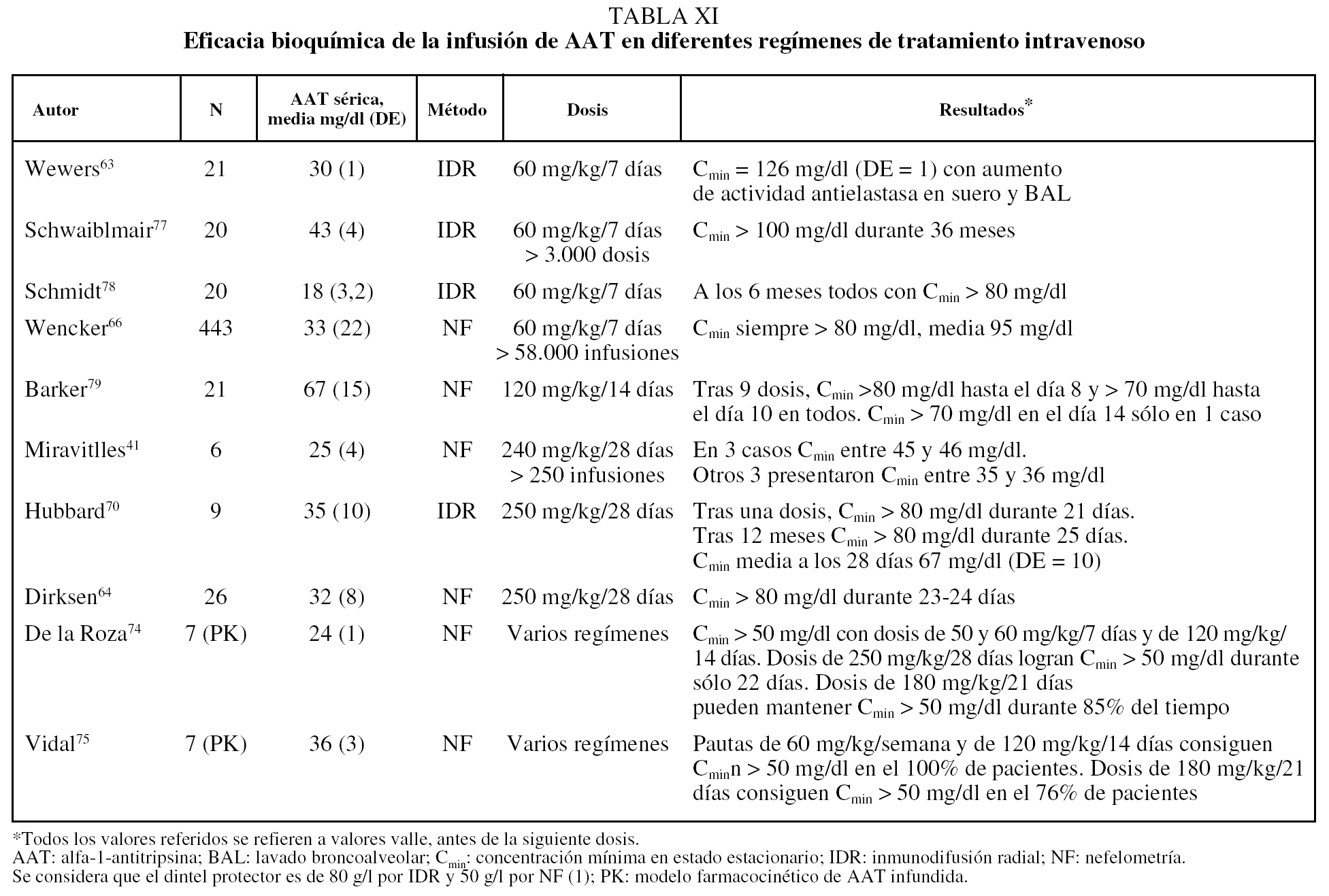
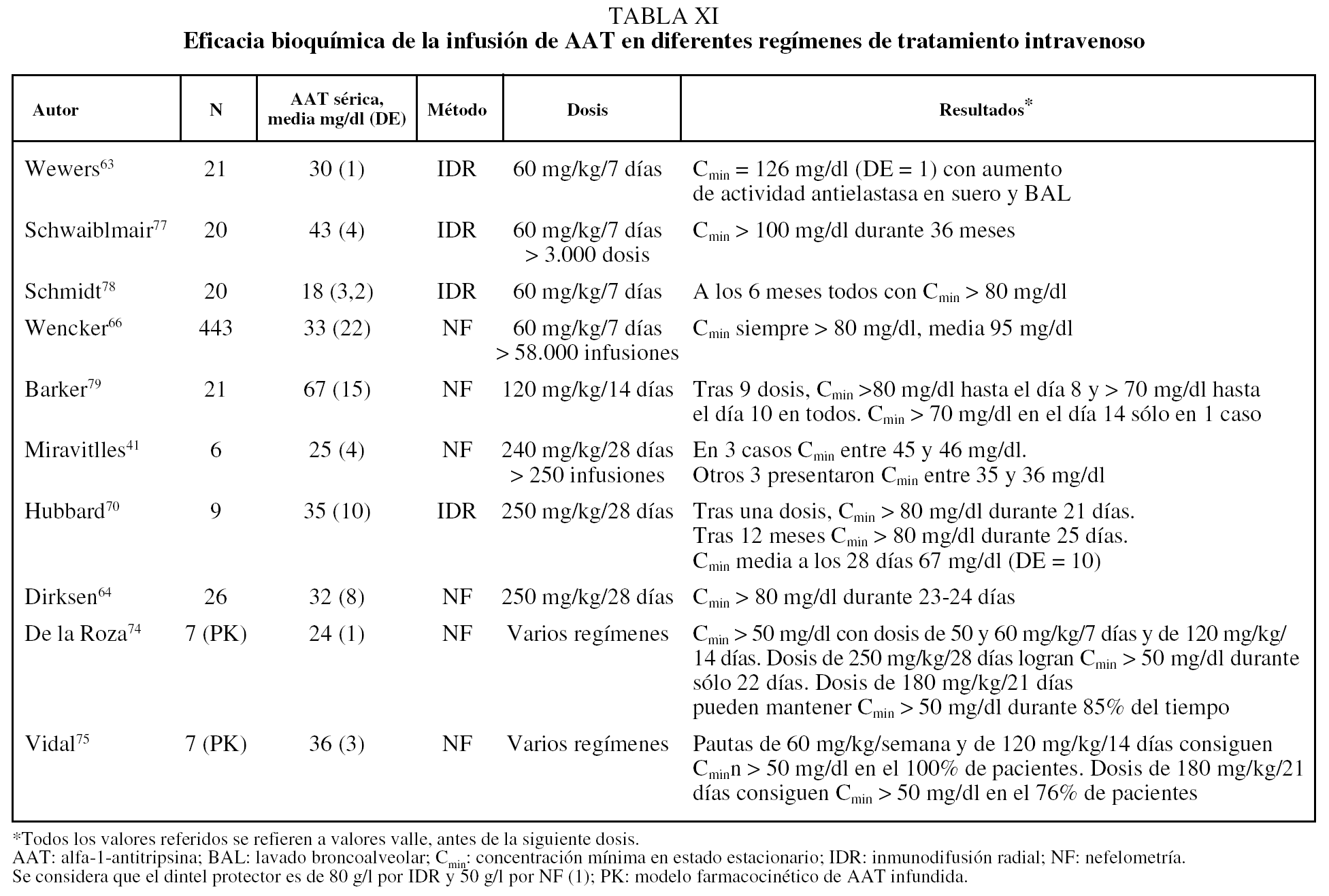
El tratamiento médico de los pacientes con enfisema por DAAT debe comprender las medidas farmacológicas y no farmacológicas comunes a los pacientes con EPOC sin DAAT61,62. Desde 1987 se dispone de AAT purificada procedente de plasma de donantes para administración intravenosa. La sustancia infundida se ha demostrado que mantiene su actividad enzimática en plasma y en lavado broncoalveolar; además su actividad pulmonar se correlaciona de forma directa con su concentración plasmática, lo que permite monitorizar el tratamiento a través de la determinación de las concentraciones plasmáticas mínimas en estado estacionario (Cmin), también llamadas concentraciones valle, que se corresponden con las concentraciones obtenidas antes de la siguiente dosis tras las necesarias para alcanzar el estado estacionario63. A partir de estudios epidemiológicos, se ha considerado que la Cmin de 0,8 g/l determinados por inmunodifusión radial o 0,5 g/l por nefelometría son capaces de proporcionar una protección adecuada al pulmón comparado con individuos normales no fumadores1. Como la vida media de la AAT infundida es de 4-5 días, las primeras pautas de administración ensayadas tuvieron una periodicidad semanal63. Aun en la actualidad, la pauta recomendada en ficha técnica es de 60 mg/kg/semana. No obstante, debido al evidente inconveniente de la administración semanal del tratamiento de por vida, se han ensayado otras pautas cada 14, 21 y 28 días. Los resultados de estas distintas pautas de administración desde el punto de vista bioquímico se recogen en la tabla XI.
Eficacia clínica
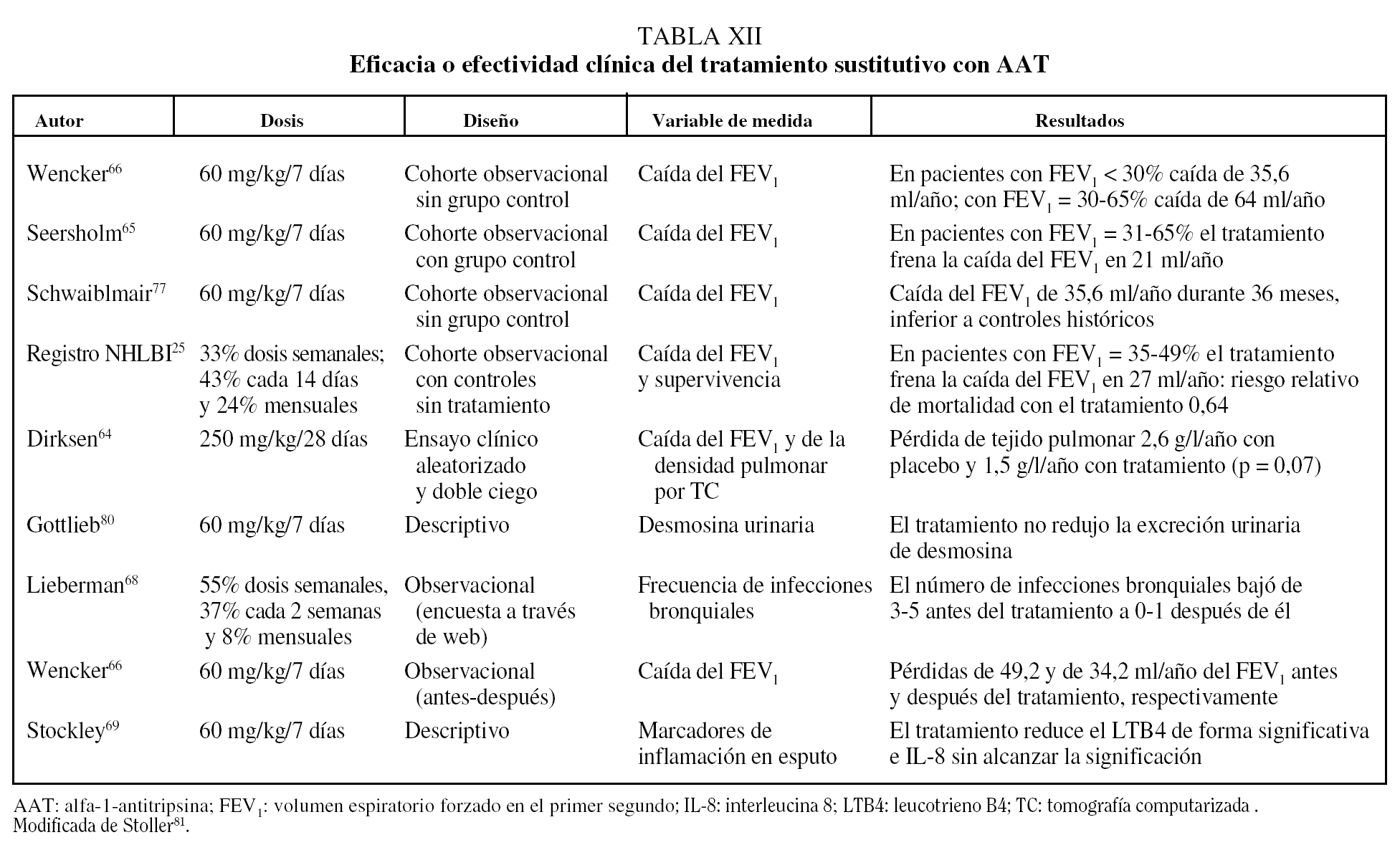
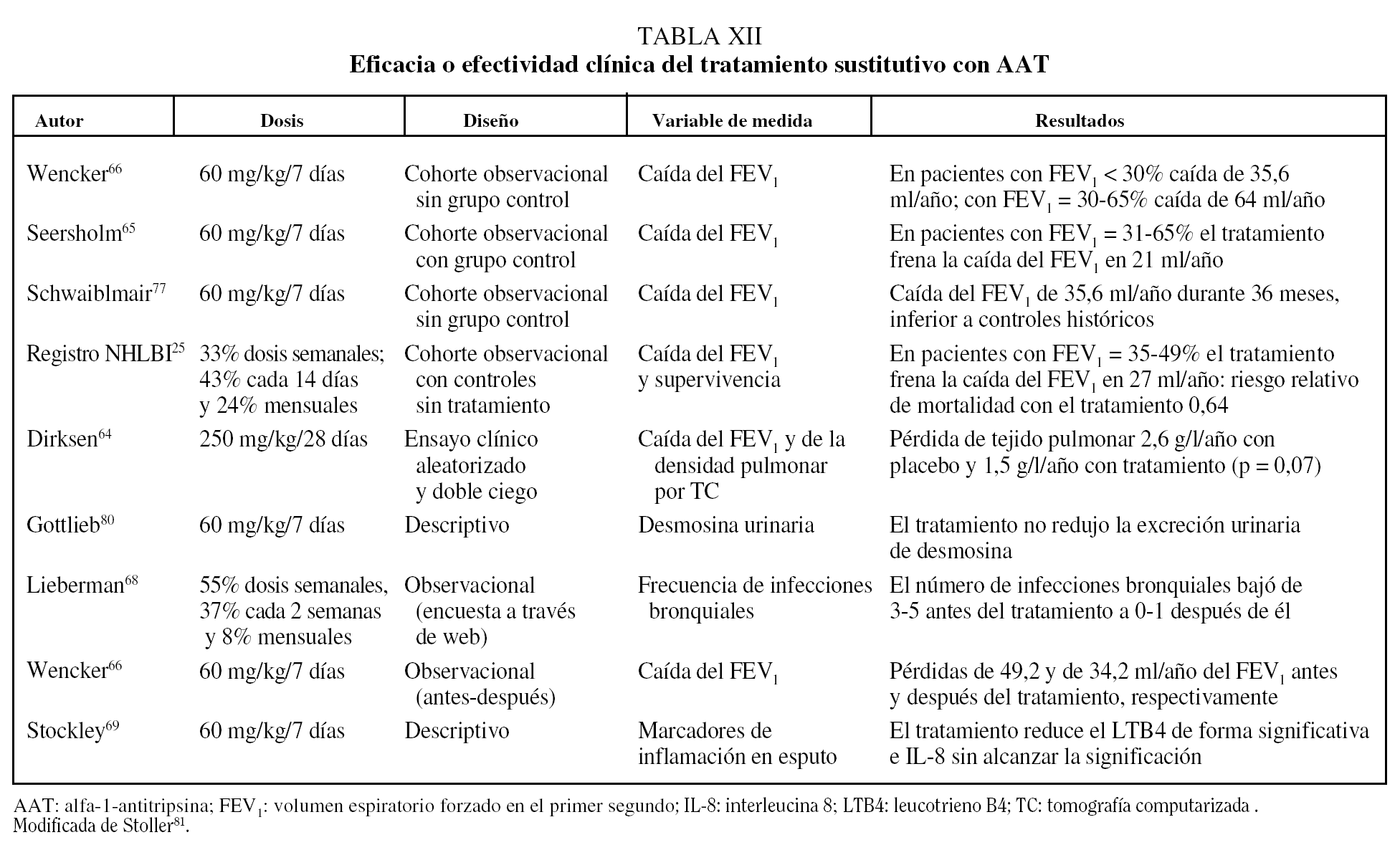
Existe un único ensayo clínico que comparó el tratamiento con AAT humana a dosis de 250 mg/kg/28 días con placebo con un diseño aleatorizado y doble ciego. El estudio incluyó a 58 pacientes tratados durante 3 años y no mostró diferencias significativas en la evolución de la función pulmonar, pero los pacientes que recibieron AAT presentaron una pérdida anual de densidad pulmonar medida por TC de 1,50 g/l comparado con 2,57 g/l en los que recibieron placebo (p = 0,07)64.
El resto de los datos que se conocen sobre la efectividad del tratamiento sustitutivo deriva de estudios comparativos de seguimiento. Estos estudios han observado una diminución significativa en la caída del FEV1 en pacientes con un FEV1 del 30-60%65,66. Además, datos del registro NHLBI en Estados Unidos sobre 1.048 pacientes seguidos durante 3,5-7 años han observado una reducción significativa del 36% en la mortalidad en los pacientes que recibieron tratamiento sustitutivo de forma continuada o intermitente comparados con los que no recibían tratamiento (p = 0,02)25.
Un efecto interesante del tratamiento sustitutivo es la posible protección frente a las infecciones bronquiales, importante también por la elevada prevalencia de bronquiectasias en esta población67. Los resultados de un trabajo observacional sugieren que los pacientes reducen la frecuencia de agudizaciones tras iniciar tratamiento sustitutivo68. Este hecho puede estar en relación con la restitución bronquial del estado de desequilibrio proteasa/antiproteasa y de inflamación en los pacientes que reciben tratamiento sustitutivo69.
El efecto del tratamiento sustitutivo en los pacientes graves (FEV1 < 30%) es difícil de observar debido a que estos pacientes fallecen o son sometidos a trasplante pulmonar antes de poder completar un seguimiento suficientemente prolongado. En el caso de los pacientes leves (FEV1 > 60%) tampoco es posible evaluar el efecto del tratamiento, ya que existe un sesgo por indicación. El escaso número de pacientes que recibe tratamiento en este estadio precoz son casos índice con síntomas especialmente graves o con una pérdida acelerada de función pulmonar, mientras que sus comparadores sin tratamiento suelen ser casos no índice que no reciben tratamiento precisamente por no tener síntomas o por mantener una función pulmonar estable25,66.
Los principales estudios que han evaluado la eficacia o efectividad del tratamiento sustitutivo se resumen en la tabla XII.
Seguridad
La infusión de AAT humana por vía intravenosa para el tratamiento crónico del enfisema por DAAT es un tratamiento que ha demostrado ser muy seguro. Las primeras experiencias se inciaron en 1987 sin observarse reacciones agudas63. Se debe destacar que no se han observado reacciones secundarias a sobrecarga de proteína tras la administración periódica a largo plazo de grandes dosis en administración a intervalos mensuales41,64,70. La mayor base de datos de efectos adversos es la del registro NHLBI, la frecuencia de efectos adversos fue de 0,02 por pac iente y mes, pero sólo un 9% se consideró grave y sólo un 1,7% requirió atención en urgencias o ingreso hospitalario. Hasta el 85% de los pacientes no presentó ningún efecto adverso. Los más frecuentes fueron cefalea (47%), vértigo (17%), náuseas (9%) y disnea (9%). No se encontró ningún caso de transmisión de hepatitis A, B, C o delta, ni VIH ni enfermedad mediada por priones. Los pacientes que recibieron infusiones semanales señalaron mayor frecuencia de efectos adversos y de efectos adversos considerados graves71.
Productos disponibles para administración intravenosa
En la actualidad disponemos en España de 2 preparados de AAT procedente de plasma humano para su administración intravenosa:
- Prolastina® (QF Bayer, S.A.) suministrada en polvo para reconstitución en viales de 50 ml. Tras la reconstitución, cada vial contiene como mínimo 20 mg/ml para una cantidad total de AAT de 1.000 mg.
- Trypsone® (Instituto Grifols, S.A.) suministrada en polvo para reconstitución en viales de 25 o 50 ml. Tras la reconstitución, cada vial contiene como mínimo 20 mg/ml para una cantidad total de AAT de 500 o 1.000 mg, respectivamente.
En el único estudio disponible, Trypsone® ha demostrado conseguir una Cmin en suero equivalente a las obtenidas con Prolastina®. También la actividad antielastasa y las concentraciones de AAT en BAL fueron equivalentes con ambos preparados72.
El tratamiento se administra en centros hospitalarios en zonas de hospital de día. La preparación del producto a infundir por la farmacia del hospital debe realizarse cuando el paciente ha llegado al centro y debe infundirse lo antes posible, ya que tras su reconstitución tiene un período de actividad de 3-4 horas. La velocidad de perfusión debe ser inferior a 0,08 ml/kg PC/min.
Criterios de tratamiento
El tratamiento sustitutivo solamente está indicado en el caso del enfisema pulmonar por DAAT. No tiene ningún efecto sobre la hepatopatía asociada al DAAT. No está documentada su posible eficacia frente a otras manifestaciones poco frecuentes del DAAT como la paniculitis.
Los criterios de tratamiento sustitutivo se basan en el reconocimiento del déficit grave de AAT, fenotipo PiZZ o variantes raras deficitarias y la demostración de enfisema pulmonar por funcionalismo32. Los casos no índice, es decir, aquellos no diagnosticados por clínica respiratoria sino por estudio familiar, deben demostrar una pérdida acelerada de función pulmonar al menos durante un año.
El tratamiento no está indicado en individuos heterocigotos PiMZ o PiSZ. Debido a que los hemoderivados pueden contener trazas de IgA, y que los pacientes con déficit de IgA pueden tener anticuerpos circulantes anti-IgA, es obligatorio descartar un déficit de IgA antes de iniciar el tratamiento. Los criterios de tratamiento se recogen en la tabla XIII.

En la actualidad no se recomienda de forma sistemática la vacunación antihepatitis A o B antes de iniciar el tratamiento1 (tabla XIV).

Pautas de administración recomendadas
No existe una única pauta de dosificación de la AAT. La ficha técnica de los diversos productos disponibles recomiendan, si no existe otra indicación médica, la dosis de 60 mg/kg/semana por ser la que primero se estudió y la mejor documentada. Sin embargo, diversos estudios han demostrado la eficacia y seguridad de otras pautas de dosificación. En efecto, en el registro NHLBI en Estados Unidos, el 67% de los pacientes ha estado recibiendo pautas diferentes de la semanal. Dadas estas experiencias, sumado a la dificultad de la mayoría de los pacientes de recibir dosis semanales de por vida y la demostración de que pautas mensuales resultaban en Cmin muy bajas41,64,70, el Registro Español recomendó la pauta de 180 mg/kg/21 días de forma general32,41. Esta pauta se ha venido utilizando en la mayoría de centros durante los últimos 10 años73.
En la actualidad, nuevos estudios de farmacocinética (PK) nos permiten conocer el comportamiento de la AAT infundida en las diversas pautas y extender las pautas recomendadas74,75. Dichas pautas se recogen en la tabla XV.

Las pautas de 50 mg/kg/7 días y 120 mg/kg/14 días han demostrado mantener Cmin superiores a las consideradas protectoras en más del 90% de los pacientes. La pauta de 180 mg/kg/21 días consigue mantener Cmin consideradas protectoras durante aproximadamente un 85% del tiempo entre dosis. En ausencia de estudios concluyentes que relacionen la eficacia clínica con medidas PK, la elección de la pauta debe ser individualizada y surgir de un compromiso entre la eficacia bioquímica, las expectativas y la disponibilidad de los pacientes y las posibilidades del centro hospitalario.
No se recomienda la determinación de las Cmin debido a que su interpretación es compleja y los ajustes de dosis posteriores requerirían un análisis PK individualizado. En caso que se sospeche que es preciso un ajuste de dosis, lo debe realizar un centro con experiencia en PK.
Otros tratamientos
Las pautas de seguimiento de los pacientes con enfisema por DAAT son similares a las de pacientes con EPOC con concentraciones normales de AAT. Estos pacientes deben recibir tratamiento de base con broncodilatadores inhalados y añadir corticoides inhalados cuando esté indicado. Se recomienda la vacunación antigripal anual y la antineumocócica, ya que se ha demostrado que presentan una buena respuesta de anticuerpos específicos67.
Los episodios de agudización se caracterizan en estos pacientes por un exceso de actividad elastasa, mucho más acusado que en pacientes con EPOC sin DAAT45. Por este motivo, el tratamiento de las agudizaciones debe ser precoz y enérgico con incremento de las dosis de broncodilatadores, corticoides orales en tandas cortas y antibióticos cuando se presenten cambios en las características o en la cantidad de la expectoración.
Se considerará la oxigenoterapia cuando se cumplan los criterios habituales. La rehabilitación respiratoria se debe ofrecer a los pacientes con alteración funcional.
A pacientes graves seleccionados se les puede ofrecer la posibilidad de trasplante pulmonar. La cirugía de reducción de volumen pulmonar ha ofrecido resultados no concluyentes en estos pacientes, ya que por las características morfológicas de su afectación pulmonar se considera que no son candidatos idóneos para esta técnica76-81.
Como norma general se recomienda una visita clínica de control semestral con espirometría simple y la determinación de volúmenes pulmonares estáticos y prueba de transferencia del CO una vez al año. La gasometría arterial y la TC de tórax se realizarán cuando haya cambios clínicos que lo justifiquen (tabla X).
*Comité del Registro Nacional de Pacientes con Déficit de Alfa-1-Antitripsina Juan Carlos Barros-Tizón, Ana Bustamante, Carlos Escudero, Pedro P. España, Maite Martínez y Francisco Rodríguez-Frías.
Colaboradores del Registro: Cristian de la Roza, Sara Vilà, Zvezda Drobnic, María Torres, Beatriz Lara, Bruno Montoro, Dolors Soy y Pilar Gispert.
Correspondencia: Dr. R. Vidal.
Servicio de Neumología. Hospital General Universitari Vall d'Hebron.
P.º Vall d'Hebron, 119-129. 08035 Barcelona. España.
Correo electrónico: ravidal@vhebron.net
Recibido: 10-2-2006; aceptado para su publicación: 11-4-2006.







