

Primary pulmonary lymphoma (PPL) accounts for less than 0.5% of primary lung neoplasms.1 Its non-specific clinical and radiological manifestations make diagnosis challenging, conferring a worse prognosis. We report a small series of cases that illustrate the most common PPL.
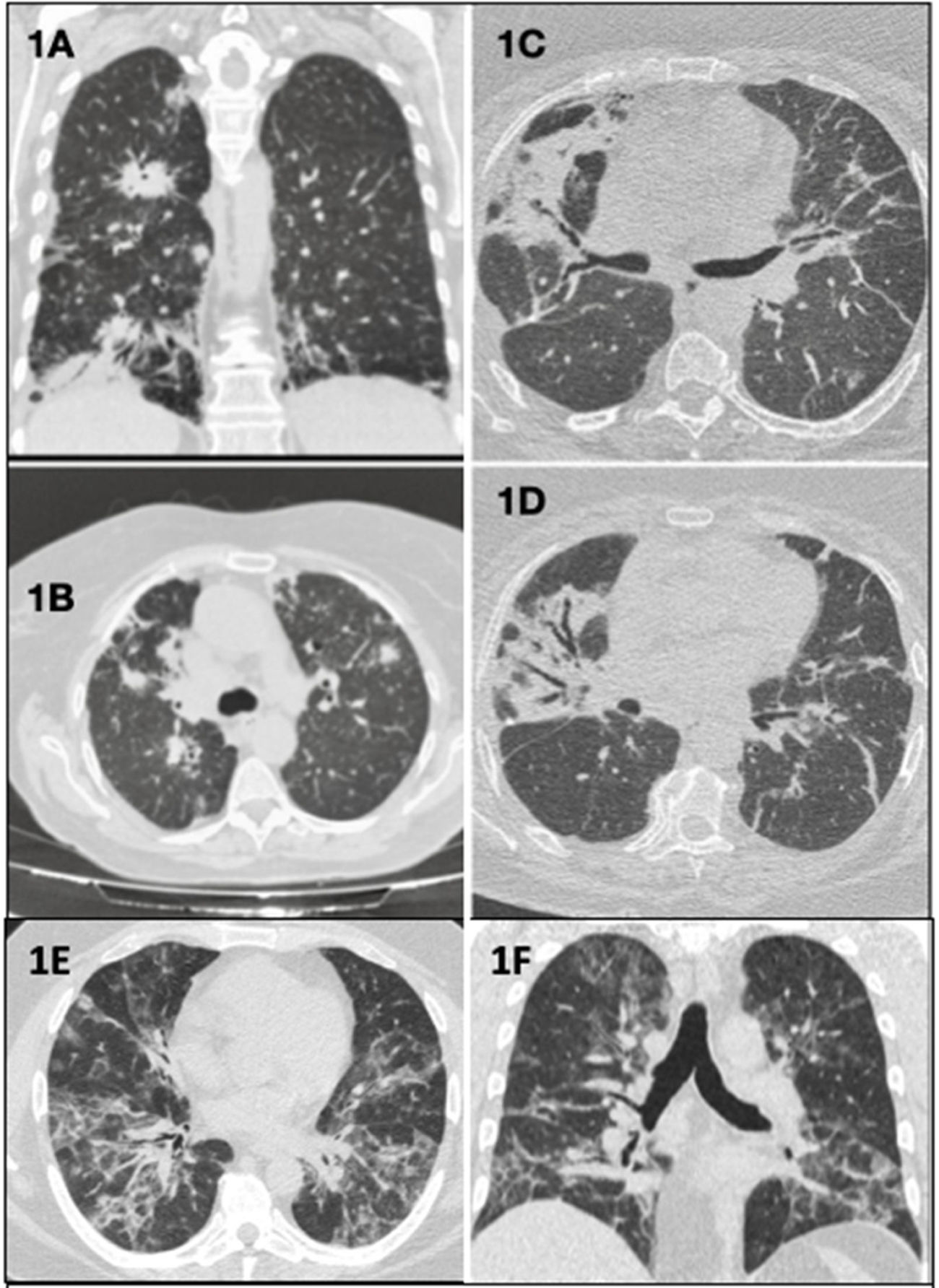
Clinical case 1: A 70-year-old woman with low-grade fever and bilateral pulmonary infiltrates on chest computed tomography (CT) (Fig. 1A and B), refractory to antibiotic and corticosteroid treatment. Surgical lung biopsy (SLB) revealed primary pulmonary diffuse large B cell lymphoma (PP-DLBCL), currently in remission after R-CHOP chemotherapy (CT) (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
 (Coronal plane) and (B) (axial plane): bilateral parenchymal consolidations with peribronchovascular, peripheral, and subpleural distribution. (C and D) (Axial planes): bilateral peribronchovascular opacities due to atelectasis and fibrosis, and bronchiectasis and traction bronchiectasis. (E) (Axial plane) and (F) (coronal plane): alveolo-interstitial opacities with bilateral and peribronchovascular distribution that coexist with other peripheral band-like ground glass opacities.")
(A) (Coronal plane) and (B) (axial plane): bilateral parenchymal consolidations with peribronchovascular, peripheral, and subpleural distribution. (C and D) (Axial planes): bilateral peribronchovascular opacities due to atelectasis and fibrosis, and bronchiectasis and traction bronchiectasis. (E) (Axial plane) and (F) (coronal plane): alveolo-interstitial opacities with bilateral and peribronchovascular distribution that coexist with other peripheral band-like ground glass opacities.
Clinical case 2: A 63-year-old woman with bilateral alveolar infiltrates (Fig. 1C and D) and peripheral blood expression of 2.3% kappa clonal B cells. SLB showed B cell infiltration, while marginal zone B cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) was confirmed on bone marrow biopsy. Pulmonary resection was performed and rituximab adjuvant therapy was administered.
Clinical case 3: A 37-year-old man with cough, skin rash, and alveolar opacities on CT scan (Fig. 1E and F). SLB showed Epstein–Barr virus (EBV)-positive atypical B cell infiltrate, establishing a diagnosis of grade 1 lymphomatoid granulomatosis (LG), and a wait-and-see approach was adopted.
PPLs are lymphoproliferative disorders resulting from clonal proliferation of B cells, that show parenchymal and/or bronchial involvement but no extrapulmonary involvement at diagnosis or up to 3 months later. They account for less than 1% of non-Hodgkin lymphomas,1 of which MALT lymphoma, PP-DLBCL, and LG are the most common.2 Their etiopathogenesis is unknown, but they are associated with autoimmune diseases,4 infections, and EBV in LG.5 Generally, they predominate in men in their 50s and 60s. Half of all patients (50%) with MALT lymphoma3 are asymptomatic at diagnosis, while PP-DLBCL and LG may have a more aggressive onset involving B symptoms.5 LG may occur with extrapulmonary involvement, the most prevalent manifestations being cutaneous, neurological and otorhinolaryngological.2 CT is often non-specific, and the most frequent findings include multiple bilateral ground glass and/or consolidative opacities.2–4
Diagnosis requires histology – the gold standard being lung biopsy obtained by CT-guided percutaneous biopsy and transbronchial biopsy, which have a sensitivity of 80% and 88%, respectively.2 Moreover, it is crucial not to postpone SLB if results are inconclusive, since erroneous diagnoses are made in 68% of cases, causing a mean delay of up to 2 years until final diagnosis.3
The lymphoid infiltrate in MALT lymphoma is CD20-, CD19- and CD79a-positive and CD5-negative, with a Ki67 of less than 10%, while PP-DLBCL infiltrate has a Ki-67 of 40%–60%. In LG, the infiltrate shows varying proportions of EBV-positive atypical B cells, determining a disease classification of grade I to III and prognostic repercussions.5
There is no specific treatment for PPL, so therapeutic management must be individualized. Different regimens (CHOP) or monotherapy with rituximab have been shown to be effective for MALT lymphoma, and the R-CHOP regimen has become standard in patients with PP-DLBCL, with good results.3 The treatment of LG, however, depends on tumor extension and histological grade, and remains controversial.5
Generally, MALT lymphomas have the best prognosis,2 followed by PP-DLBCL, while LG has a 5-year mortality rate of 63%–90%.5
In conclusion, the non-specific presentation of PPL is a common source of diagnostic error, so it must be included in the differential diagnosis. Immunohistochemical study is essential for early identification and treatment.







