




Respiratory tract affectation is frequent in some types of vasculitis, fundamentally in those associated with anti-neutrophil cytoplasmic antibodies (ANCA). The clinical, radiological and histopathological presentation is also heterogeneous and conditions the evolution. It is therefore necessary to establish an early diagnosis based on the symptoms because, thanks to new treatments, and despite them being potentially serious diseases, their prognosis has improved considerably in recent years.
The present paper updates the diagnosis and the new therapeutic options for pulmonary vasculitis.
La afectación del tracto respiratorio es frecuente en algunos tipos de vasculitis, fundamentalmente en las asociadas a anticuerpos anticitoplasma de neutrófilo. La presentación clínica, radiológica e histopatológica también es heterogénea y condiciona la evolución. Es necesaria, por tanto, una orientación clínica y diagnóstica precoz ya que, gracias a los nuevos tratamientos, y a pesar de ser enfermedades potencialmente graves, su pronóstico ha mejorado de manera considerable en los últimos años.
En el presente trabajo se realiza una actualización del diagnóstico y las nuevas opciones terapéuticas de las vasculitis pulmonares.
The term “systemic vasculitis” represents a very heterogeneous group of diseases that are characterized by inflammation of the blood vessel walls. An inflammatory infiltrate has been described, which can be either acute or chronic and entails destructuring of the blood vessels with reduced blood flow to the tissues.1,2 One or several organs or systems may be affected, depending on the size and location of the affected vessels, and the clinical presentation may therefore be proteiform.
Historically, the vasculitides have been classified according to their histopathology (Figs. 1 and 2).3 At the Chapel Hill conference,4 nomenclatures and definitions for the different types of systemic vasculitis were proposed (Table 1). This classification is based on the size of the vessels involved and it incorporates the existence of ANCA. The large-cell vasculitis, polyarteritis nodosa and Kawasaki disease included in said classification rarely present lung affectation.4,5 In contrast, other vasculitides that are not included in the Chapel Hill classification but do have lung affectation are those associated with autoimmune diseases such as Behçet's disease or systemic erythematous lupus. The Chapel Hill consensus is still valid though, because it offers a useful framework for the classification of vasculitides according to the size of the damaged vessels, which provides an approach for the different clinicopathological presentations.
.")
Classification of vasculitides. ANCA: anti-neutrophil cytoplasmic antibodies; IF: immunofluorescence (taken from Gómez-Román3).
. ANCA: anti-neutrophil cytoplasmic antibodies; IF: immunofluorescence (taken from Gómez-Román5).")
Classification of Systemic Vasculitides According to the Chapel Hill Classification.
| Large-vessel vasculitis |
| Giant-cell arteritis |
| Takayasu arteritis |
| Medium-vessel vasculitis |
| Polyarteritis nodosa |
| Kawasaki disease |
| Small-vessel vasculitis |
| Wegener's granulomatosisa |
| Churg-Strauss syndromea |
| Microscopic polyangiitisa |
| Schönlein-Henoch purpura |
| Cryoglobulinemia |
| Cutaneous leukocytoclastic angiitis |
The respiratory tract may be affected in systemic vasculitis, although this is more frequent in ANCA-associated vasculitis, as we will see below.
Types of Lung Affectation in VasculitisPulmonary vasculitides are a heterogeneous group of diseases that are very different in their clinical and radiological presentation, and they are frequently associated with the presence of systemic affectation.6 Despite including such different entities, there are common symptoms and signs that are suggestive and that, together with biological data, provide a more specific differential diagnosis.7–9
The small blood vessels of the lungs may be affected (pulmonary capillaritis) and cause diffuse alveolar hemorrhage, characterized by the triad of diffuse alveolar infiltrates, hemoptysis and fall in hematocrits and/or hemoglobin.10 Also associated with some types of pulmonary vasculitis (vasculitis ANCA) is the presence on imaging tests of cavities or nodules as well as inflammation of the large lung blood vessels, which entail the appearance of aneurisms and thrombosis.11
Diffuse Alveolar HemorrhageThe pulmonary affectation of vasculitis may develop in any blood vessel, regardless of size. Vasculitis in the lung microvasculature is known as pulmonary capillaritis. Although the diagnosis is pathologic, its presence makes it necessary to rule out underlying disease12 because it may be the first manifestation, as is frequently found, in vasculitis that presents ANCA or systemic autoimmune diseases. The most serious complication of lung capillaritis that is a resulting consequence of the damaged microcirculation is pulmonary hemorrhage (PH). Although the pulmonary alveolar hemorrhage may be due to diffuse alveolar damage caused by multiple etiologies, in the case of vasculitis it is a consequence of the inflammation of the lung capillaries themselves.13
It is characterized by the presence of an interstitial infiltrate with neutrophils and fibrinoid necrosis in the capillary walls.14 Secondary to this inflammation and to the presence of the neutrophils, there is a loss in the integrity of the basement membrane of the alveolocapillary union with disruption and extravasation of red blood cells into the alveolar spaces. The presence of hemosiderophages in the bronchoalveolar lavage is very typical as the alveolar macrophages phagocyte the erythrocytes that flow through the lesion in the basement membrane of the vascular endothelium to the alveolus.15
PH has been described as the most severe manifestation of the vasculitides associated with ANCA and it is more frequent in microscopic polyangiitis (MPA)8,16 and Wegener's granulomatosis.17–20 Less frequently, it has also been associated with Goodpasture syndrome,21 Churg-Strauss syndrome,22–25 Schönlein-Henoch purpura,26–28 Takayasu's arteritis, giant-cell arteritis, cryoglobulinemia, polyarteritis nodosa or Beçhet's disease29,30 (Table 2).31 From a clinical standpoint, it is defined as a syndrome that is characterized by the presence of hemoptysis (even though it may not be present in one-third of cases32), anemia, acute respiratory insufficiency and a sudden onset of alveolar infiltrates.33 It is a potentially fatal complication with an unpredictable clinical evolution. Likewise, other more non-specific symptoms may appear, such as thoracic pain, cough, fever or general syndrome for weeks or months prior.
Lung Affectation and Vasculitis.
| Vasculitis with frequent lung affectation | Churg-Strauss diseaseWegener's diseaseMicroscopic polyangiitis |
| Vasculitis with infrequent lung affectation | Schönlein-Henoch purpuraTakayasu arteritisGiant-cell arteritisCryoglobulinemiaPanarteritis nodosaBeçhet's disease |
Adapted from Fishbein.31
Chest radiography is nonspecific, shows either focal or diffuse alveolar infiltrates that on many occasions require performing thoracic high-resolution computed tomography in order to define and confirm the findings. Respiratory function tests are characterized by an increased carbon monoxide diffusion capacity (DLCO), although occasionally this is not feasible if the patient is in critical condition.34 Bronchoscopy with bronchoalveolar lavage is required, if the clinical situation of the patient allows, as it helps establish the diagnosis and rule out other entities. The presence of more than 20% of hemosiderophages in the bronchoalveolar lavage is diagnostic for pulmonary alveolar hemorrhage, even in those cases with subclinical presentation.
The treatment of patients with pulmonary alveolar hemorrhage depends on the severity of the hemorrhage as well as the extrapulmonary manifestations of the vasculitis. In severe cases, it may be necessary to transfer the patient to an intensive care unit with respiratory support.
Different classification systems have been proposed depending on severity, and the intensity of treatment depends on this severity. In all cases, an induction treatment is used to try to control the activity of the vasculitis and reach a state of remission in a few weeks, followed by a maintenance phase in order to prevent relapse.35
The induction treatment in those cases in which the patient presents stability and does not require ventilatory support is based on regimes of combined therapy that include immunosuppressants like glucocorticoids and cyclophosphamide, methotrexate36 or rituximab.37,38 These last two drugs have been shown to be effective and to have less side effects than the first; however, more treatment time is necessary to achieve remission. Rituximab has recently been proposed as an alternative to cyclophosphamide because similar effectiveness rate have been demonstrated with the two immunosuppressants.39 In cases with severe affectation, in addition to the previously described treatment, plasmapheresis has been proposed as a therapeutic option. Nevertheless, there is no solid scientific evidence to support this. For cases that are refractory to the previous treatment, there have been assays with the use of immunoglobulin and rituximab that have shown good results.40,41
In the maintenance treatment, low doses of glucocorticoids have been used as they have been shown to prevent relapses.42 In addition, in order to be able to maintain low doses, cyclophosphamide can be substituted for another immunosuppressant after 3–6 months of treatment, such as methotrexate or azathioprine.43 Other treatments such as mycophenolate mofetil or leflunomide have been demonstrated to be less efficient than azathioprine in order to prevent relapses.44
Granulomatous Lung DiseaseVasculitides that cause pulmonary granulomatous disease are characterized by the presence of ANCA-type antibodies. They produce granulomatous inflammation of both the upper and lower respiratory tract with the presence of necrotizing vasculitis and renal affectation with hematuria and proteinuria. In most cases, the antibodies are directed against proteinase 3-antineutrophil cytoplasmic antibody (PR3-ANCA).45 In the pathogenesis of this type of lung affectation, an inflammatory cell response has been implicated that results in the formation of granulomas made up of an inflammatory infiltrate comprising neutrophils, lymphocytes, plasma cells, histiocytes and eosinophils.18 Likewise, the necrosis of the pulmonary parenchyma itself can lead to the formation of micro-abscesses or areas with a necrotic center surrounded by a ring of histiocytes that offer a typical radiological image (halo sign). There are also reports of increased bronchovascular markings in the parenchyma, hilar lymphadenopathies and associated pleural effusion.46 When this type of lesions appears related with a phase of activity of the disease, the inflammatory lesions are active and respond properly to treatment with immunosuppressants.47 With said treatment, the cavitating nodules and those larger than 3cm, as well as the infiltrates, tend to disappear and leave small lines of cicatricial fibrosis.48 The treatment in this type of affectation is similar to that previously described for capillaritis with alveolar hemorrhage.
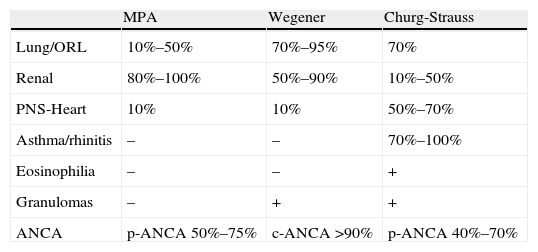
Vasculitis With Lung AffectationThe lungs are possible target organs of systemic vasculitides, and in some, like those associated with ANCA, lung affectation is very frequent (Table 3). The term “pulmonary vasculitis” does not refer to just one entity, but instead to the clinical-pathological process that takes place in the genesis of the pulmonary manifestations of these diseases. In Spain, the five-year incidence has been described for some of these vasculitides: 2.95cases/million inhabitants for Wegener's disease, 7.91cases/million inhabitants for MPA and 1.31cases/million inhabitants for Churg-Strauss disease.49
Characteristics of Vasculitides Associated Anti-neutrophil Cytoplasmic Antibodies.
| MPA | Wegener | Churg-Strauss | |
| Lung/ORL | 10%–50% | 70%–95% | 70% |
| Renal | 80%–100% | 50%–90% | 10%–50% |
| PNS-Heart | 10% | 10% | 50%–70% |
| Asthma/rhinitis | – | – | 70%–100% |
| Eosinophilia | – | – | + |
| Granulomas | – | + | + |
| ANCA | p-ANCA 50%–75% | c-ANCA >90% | p-ANCA 40%–70% |
ORL: otorhinolaryngological affectation; PNS: peripheral nervous system; ANCA: anti-neutrophil cytoplasmic antibodies.
Wegener's granulomatosis, currently known as granulomatosis with polyangiitis, is a systemic vasculitis characterized by granulomatous inflammation of the airway with necrotizing vasculitis in the small-caliber blood vessels. In this disease, there may be other types of coexisting affectations, for instance renal or nervous, like in other vasculitides. The presence of necrotizing glomerulonephritis is typical.4
Respiratory tract affectation is rather frequent, in the upper as well as the lower tract.50,51 In the upper airway, a sensation of nasal obstruction is frequent, along with rhinorrhea and ulcers, which in some cases cause perforation of the septum. The clinical manifestations due to lower tract affectation include cough, dyspnea and/or hemoptysis with pleuritic chest pain. There are reports of tracheobronchial stenosis that can condition the presence of dyspnea and stridor in some patients.52 There may also be pleural effusion, which is usually unilateral.53 One frequent complication of Wegener's disease is pulmonary alveolar hemorrhage due to capillaritis, which has been previously reported in a high percentage of patients.32 The clinical presentation may be subacute or acute with dyspnea, hemoptysis and the presence on chest radiograph of recent-onset bilateral alveolar infiltrates, and at the same time a fall in hematocrit is detected. As has been indicated, there is a characteristic increase in the CO diffusing capacity as well as the presence of siderophages in the bronchoalveolar lavage.54 In this vasculitis, there are typically multiple lung nodules, which are occasionally cavitating,55 as well as signs of bronchial obstruction, pulmonary infiltrates or parenchymal fibrosis. The lung nodules occasionally present the halo sign with a surrounding ring of ground-glass opacity,56 which is characteristic. Radiology may help determine whether the disease is in an active phase. The presence of ground glass pattern together with lung nodules greater than 3cm is indicative of the disease's activity.57 Diagnosis is not always simple because, although the radiological findings are evident, the performance of transbronchial biopsy58 or cytology of the bronchoalveolar lavage is poor. The best method for diagnosis is open biopsy. Given the presence of pulmonary infiltrates or nodules, it is possible to find necrotizing granulomas and vasculitis in up to 90% of cases.59 Although many parenchymal lesions improve after treatment, it is possible to find residual cicatricial lesions in most patients.
Wegener's disease is characterized by an associated presence of ANCA that, at the same time, are used to monitor response to treatment.60 High c-ANCA titers have been reported in more than 90% of the patients with active generalized disease, and from 40 to 70% of the patients with local active disease.60,61 More than 90% of the patients with Wegener's disease present with ANCA against proteinase 3 and the remaining 10% against myeloperoxidase.62 The change in the titers of these antibodies correlates with the disease activity. Nevertheless, they may remain high in 40% of the patients after reaching clinical remission.
Microscopic PolyangiitisMPA is a non-granulomatous small-vessel vasculitis associated with the presence of ANCA. Renal affectation is very frequent in the form of necrotizing glomerulonephritis, as well as respiratory affectation, either simultaneous or isolated.63 It is part of the pulmonary-renal syndromes, like Wegener's granulomatosis, and it may debut this way or develop it over the course of its evolution.64 The most frequent lung presentation is alveolar hemorrhage due to capillaritis with a prevalence of around 15%–30%,65 depending on the series. The evolution of diffuse alveolar hemorrhage progresses rapidly in most patients, although a minority may have a more insidious beginning. The symptoms may precede the diagnosis in one out of every four patients.63 MPA may simultaneously affect not only other organs or systems, like the nervous or muscular-skeletal systems, but also the gastrointestinal tract or heart. Therapeutic decisions should be made according to the severity and the pattern of organ affectation.
Churg-Strauss DiseaseChurg-Strauss disease, also known as allergic granulomatosis with angiitis, is a syndrome characterized by granulomatous inflammation with abundant eosinophils and necrotizing vasculitis that affects small and medium-sized vessels, fundamentally of the lungs,66 that are associated with asthma and eosinophilia.19
The evolutive course of the disease usually presents several phases. Initially, there is typically the presence of allergic asthma with rhinitis and occasionally sinusitis and nasal polyposis. Afterwards, eosinophilia appears in the peripheral blood, and finally there are manifestations of systemic vasculitis. The presence of asthma is almost constant and present in 95% of the patients.67 As it is a disease that occurs in different phases, asthma may precede the vasculitis by several years.68 In most cases, the asthma remits after the vasculitis, but it may persist years later, requiring corticosteroid treatment for long period of time or other additional drugs in order to be able to reduce the dosage of corticoids.69,70 Other less frequent lung manifestations are diffuse alveolar hemorrhage71 and the presence of pleural effusion.72 Likewise, it may affect other organs besides the lungs, such as the nervous system, cardiovascular system, skin73 or muscle and skin the form of polymyositis.74
In the etiopathogenesis of the disease, the presence of immune complexes in the vascular walls had been implicated in the past, but more recently it has been seen that there is an underlying vasculitis mediated by ANCA.75
The histopathologic presentation in the lungs may combine the presence of extravascular granulomas, vasculitis and eosinophilic pneumonia76; however, the coexistence of the three lesions is uncommon. Granulomas are made up of a center of eosinophils surrounded by an infiltrate of histiocytes, and vasculitis is also characterized by an infiltrate of eosinophils in the media and intimal lining of the blood vessels.
Chest radiography demonstrates the presence of patchy evanescent pulmonary infiltrates without a clear lobar distribution, as well as lung nodules, which are rarely cavitating, or other alterations such as septal thickening, ground glass pattern or pleural effusion.77
Laboratory data support the clinical diagnosis in the presence of leukocytosis with marked eosinophilia, increased acute phase reactants and IgE, which frequently correlates with the activity of the disease.68 In Churg-Strauss disease, ANCA are positive in 40%–60% of cases.78 The pattern is usually p-ANCA, in contrast with Wegener's granulomatosis. With treatment, there is an observed increase in survival of these patients.79
Other VasculitisPolyarteritis Nudosa (Classic PAN)Polyarteritis nudosa is a vasculitis of medium-sized arteries, arterioles, capillaries or venules. The affectation of the respiratory system is uncommon.80 The presence of pulmonary alveolar hemorrhage has been described in some cases of PAN associated with hepatitis B virus, as well as diffuse interstitial infiltrates.81–83 There are also documented cases with pulmonary nodules not associated with ANCA.84 Pulmonary arteries are rarely affected, unlike the bronchial arteries. Previous studies about the lung affectation of this vasculitis81 reported bronchial artery lesions with alveolar damage along with data for fibrosis.
CryoglobulinemiaCryoglobulinemia is a small vessel vasculitis that is associated with the presence in serum of cryoglobulins and in many cases hepatitis C virus. The most frequent affectation is cutaneous, followed by renal in the form of glomerulonephritis.4 Lung affectation, when present, is usually mild. Patients present with cough, exertion dyspnea or pleuritic chest pain.85 Although less common, the development of alveolar hemorrhage as a consequence of pulmonary capillaritis has been described.86–88
On imaging tests, it may not be possible to make any findings, or contrarily there may be data of interstitial fibrosis,89 pleural effusion or lung infiltrates.90 In respiratory function testing, all these findings translate into a restrictive affectation with reduced CO diffusion.91 The bronchoalveolar lavage of these patients shows a higher number of T lymphocytes as a consequence of the underlying alveolitis.
Schönlein-HenochSchönlein-Henoch disease is a small blood vessel vasculitis that is a consequence of the deposition of IgA immune complexes. It is a disease that is typical of childhood, although cases have been described in adults.92 The most common affectations are cutaneous, renal and intestinal. Lung affectation is very rare, although cases of usual interstitial pneumonia have been described, as well as cases of more severe pulmonary alveolar hemorrhage.93,94 As in other vasculitis, in the presence of alveolar damage, there have been reports of DLCO alterations, which is higher in cases of alveolar hemorrhage. In addition to the necrosis of the pulmonary capillary walls, histopathology reports have also revealed deposition of IgA in the alveoli proximal to the areas of capillaritis.92
Giant-cell ArteritisGiant-cell arteritis is a granulomatous vasculitis that typically affects large vessels like the aorta and its branches. Its presentation is characteristic of advanced age and it is frequently associated with the presence of rheumatic polymyalgia.
Lung affectation is uncommon.95 It may also present with general symptoms such as fever and asthenia, dry cough,96 pleuritic chest pain, dyspnea97 and occasionally pleural effusion or interstitial lung disease.98,99 Radiologically, the presentation is variable. It may oscillate between interstitial infiltrates, lung nodules or, in the most severe cases, alveolar infiltrates as a consequence of lung hemorrhage.100–102 In any case, the most characteristic affectation of this vasculitis is vascular aneurisms that may be detected by means of imaging tests, which, in cases of dissection, have a high mortality.103 The histopathology of this disease shows data of vasculitis with inflammation in the adventitia and the media of large blood vessels with presence of giant cells, which conditions a fibrinoid necrosis with destruction of the vascular wall.104
Takayasu's ArteritisTakayasu's arteritis is a granulomatous vasculitis that affects large blood vessels like giant-cell arteritis. Unlike the former form, Takayasu predominantly affects young people. Physical examination in this disease is crucial as it may be suspected in a young patient with arterial hypertension and asymmetry of the peripheral pulses on examination.105
The lung affectation is usually subclinical, although in cases with symptoms, cough and exertion dyspnea are typical initial manifestations.106 There are few cases that have reported the presence of PH due to capillaritis, pulmonary hypertension or micro aneurism rupture.107–109 Other less frequent lung manifestations have been described, such as pleural effusion, pulmonary infiltrates or pulmonary hemorrage.110
Histopathology shows an inflammatory infiltrate made up of mononucleated cells and giant cells that generate granulomas and condition the fragmentation of the media, elastic lamina and therefore, the vascular wall. Later, thrombotic phenomena may appear with obliteration of the lumen of blood vessels and fibrosis.111 As in other vasculitis such as giant cell, magnetic resonance112 and positron emission tomography (PET) have been demonstrated to be very useful in the diagnosis, as well as for monitoring the activity of the disease. Specifically, PET is able to demonstrate the degree of inflammation and its extension with high sensitivity.113
TreatmentThe treatment of vasculitis with lung affectation usually includes the use of corticosteroids and other immunosuppressants. It must be kept in mind that any treatment of this type has potential adverse effects, so they should be used depending on the degree of affectation of the disease and thus avoid possible toxicities.
In order to classify the degree of activity of these diseases, several systems have been proposed. The European Vasculitis Study (EUVAS) Group has proposed a classification based on five levels: localized disease, early generalized disease, generalized disease, severe disease and refractory disease. Logically, more aggressive treatments are used when the degree of activity or severity of the disease is greater. After the introduction of immunosuppressant treatment, the 5-year mortality of the patients with vasculitis has gone down from 50% to 12% since the 1970s.50
Induction TreatmentThe first line of induction treatment that is usually used is corticosteroids associated with another immunosuppressant, almost always cyclophosphamide. This drug is used at a dosage of 2–3mg/kg/day,114,115 which may be adjusted depending on the appearance of adverse effects or on the patient response. In recent decades, several studies have demonstrated a benefit in the application of this drug in pulses every 3–4 weeks.116
As an alternative to cyclophosphamide, other drugs like methotrexate have been assayed. Methotrexate has also been shown to be a good induction treatment, although it may not be indicated in patients with renal affectation due to vasculitis. Methotrexate has been demonstrated to be as effective as cyclophosphamide for induction, but at the same time a higher number of relapses have been reported after one year of treatment.36
Maintenance TreatmentOnce remission is reached, there are several options for maintenance treatment. Azathioprine and methotrexate have demonstrated to be equivalent117 both in efficacy as well as in the relapse rates of the disease. Another therapeutic option is mycophenolate mofetil, which, although it has been demonstrated to be a good inductor drug, in a study comparing it with azathioprine, it was not shown to be superior118 and in contrast, it did present higher relapse rates than azathioprine.
PlasmapheresisPlasmapheresis was introduced for the first time in the 1970s in order to eliminate circulating immune complexes in the disease due to anti-glomerular basement membrane antibodies.119
The objective of this technique is based on the discovery of circulating antibodies (ANCA) associated with vasculitis that produce kidney and lung damage. In addition to the withdrawal from the systemic circulation of said antibodies, other beneficial aspects of this technique have been seen, such as the withdrawal of other factors like complement, fibrinogen, immunoglobulins, adhesion molecules and replacement capacity of clotting factors. In addition, it has been observed that the renal function of these patients can be improved, avoiding the need for dialysis.
There have been studies done in the different types of vasculitis. In patients with Wegener's granulomatosis, better renal function has been demonstrated in those subjected to plasmapheresis120 as well as an increase in survival associated with renal causes. In patients with MPA, there are few data, although some recent studies have shown a faster induction to remission in patients with plasmapheresis associated with immunosuppression.121
Where there is more evidence about the use of plasmapheresis is in the cases of pulmonary alveolar hemorrhage, although there have been no controlled clinical assays. Some contradictory data have been reported. Some studies showed good results with resolution of the diffuse alveolar hemorrhage in 100% of the patients, although one-third of them did not present renal affectation due to vasculitis.122 In another study, however, the treatment with plasmapheresis was associated with a mortality rate of 50% after two years.123
With all of this, plasmapheresis could be an adjuvant treatment to immunosuppression during acute phase in those vasculitides with moderate-severe affectation, as well as in those cases of refractory disease. A combination is recommended of plasmapheresis, corticosteroids and cyclophosphamide in the treatment of disease with severe affectation after the data provided by the MEPEX study124 developed in patients with ANCA vasculitis and renal affectation. The patients with plasmapheresis (69%) presented a lower rate of mortality and less need for dialysis.
Inhibitors of Tumor Necrosis Factor (TNF-α)Tumor necrosis factor-alpha is overexpressed in vasculitides. Due to this, there have been assays of biological drug treatment compared with this receptor. Infliximab, approved for the treatment of other autoimmune diseases, has demonstrated good results for inducing remission in patients with refractory disease.125,126 Nevertheless, it has not been seen to be an efficient treatment for mid- or long-term maintenance.127
There have been few studies done about TNF-α, and more clinical experience is necessary. In a study with adalimumab, it was seen that lower levels of corticosteroids were required in the joint treatment with cyclophosphamide in patients with ANCA vasculitis and severe affectation.128 With this group of treatments, paradoxically, there have been recent reports of the appearance of some cases of vasculitis secondary to medication.129
Intravenous ImmunoglobulinIntravenous immunoglobulin has been used as treatment, either alone or after previous standard treatment, for those cases of vasculitis with persistent disease, in order to maintain remission. Their effect is limited over time and could be useful when there is contraindication for other therapies.130 There are studies with good results in ANCA vasculitis, with remission reached in around 50% of cases in combination with corticosteroids and/or other immunosuppressants.131,132 More recent studies offer good data with remission rates of 60% in the monthly administration of immunoglobulin together with standard therapy in vasculitis with relapsing disease.133 In the light of these results, and given the good tolerance observed, the use of intravenous immunoglobulin together with corticosteroid treatment and immunosuppressants, may be recommended in patients with relapse in their vasculitis.60
Anti-CD20 Monoclonal Antibody (Rituximab)B-lymphocytes play an important role in the pathogenesis of autoimmune diseases, including vasculitides with ANCA.134 It is known that the number of circulating lymphocytes correlates with the activity of the disease.
Rituximab is a monoclonal antibody against the CD20 antigen expressed on the surface of B lymphocytes. There are few studies about their use in vasculitis, but they show good results.135 It has been used as induction therapy compared with cyclophosphamide in positive ANCA vasculitides, demonstrating that it is not inferior, including in patients with alveolar hemorrage136 and it has demonstrated efficacy and few secondary effects when used alone.137,138 These positive clinical data mean that this treatment currently represents one of the main clinical study pathways for the treatment of these diseases.
ConclusionRespiratory tract affectation is frequent in some types of vasculitides, fundamentally in those associated with ANCA. The clinical, radiological and histopathological presentations are also variable and condition evolution. A clinical orientation and early diagnosis are therefore necessary because, thanks to new treatments and despite being potentially severe diseases, the prognosis of these patients has improved considerably.138
Conflict of InterestsThe authors declare no conflict of interest.
Please cite this article as: Martín-Suñé N, Ríos-Blanco JJ. Afectación pulmonar de las vasculitis. Arch Bronconeumol. 2012;48:410-8.







