


En los últimos 10 años se ha constatado un creciente interés por el estudio de la inmunidad innata, particularmente por el posible papel que los denominados “receptores toll-like” (TLR) pueden desempeñar en la patogenia de algunas enfermedades respiratorias, como, por ejemplo, el asma, la enfermedad pulmonar obstructiva crónica y las infecciones. Los TLR son una familia de proteínas transmembranarias de tipo I, responsables del reconocimiento de patrones moleculares asociados a patógenos (PAMP, de pathogen-associated molecular patterns), y expresados por un amplio espectro de agentes infecciosos. Este reconocimiento lleva a una rápida producción de citocinas y quimiocinas, lo que proporciona una respuesta adaptativa duradera contra el patógeno. En la actualidad se considera que la administración de fármacos que modulen, al alza o a la baja, la actividad de estos receptores puede suponer un gran avance terapéutico en el manejo de dichas enfermedades.
El propósito de la presente revisión es describir los diferentes TLR, definir su posible papel en la patogenia de las principales enfermedades respiratorias y, finalmente, conjeturar las posibilidades terapéuticas que su modulación, agonista o antagonista, ofrece como posibles dianas terapéuticas.
There has been growing interest in the last 10 years in the study of innate immunity, in particular because of the possible role that toll-like receptors (TLR) may play in the pathogenesis of some respiratory disease such as for example, asthma, chronic obstructive pulmonary disease, and infections. TLR are a family of type 1 transmembrane proteins, responsible for recognizing molecular patterns associated with pathogens (PAMP, pathogen-associated molecular patterns), and expressed by a broad spectrum of infectious agents. This recognition leads to quick production of cytokines and chemokines which provides a long-lasting adaptive response to the pathogen. Currently, it is considered that the administration of drugs which modulate the activity of these receptors upwards or downwards may represent major therapeutic progress for handling these diseases.
The aim of this review is to describe the different TLS, define their possible role in the pathogenesis of the main respiratory diseases and finally, speculate over the therapeutic possibilities which their modulation, agonist or antagonist, offers as possible therapeutic targets.
El sistema inmunitario consta de varias líneas de defensa principales1. La inmunidad innata (natural o inespecífica), que carece de especificidad y de memoria, constituye la primera línea de defensa del organismo, sus componentes están siempre presentes y dispuestos para actuar inmediatamente, sin requerir un tiempo de latencia para desencadenar una respuesta. La inmunidad adquirida (adaptativa o específica), también conocida como respuesta inmunitaria, es mucho más compleja que la inespecífica y se caracteriza por la adaptabilidad al antígeno, la especificidad y la memoria. Esta inmunidad adquirida identifica péptidos específicos de patógenos presentados por células presentadoras de antígenos, las cuales, a su vez, activan una respuesta inmunitaria celular y humoral, mediada por células T (celular) y B (humoral)2. Una respuesta inmunitaria eficiente dependerá de la interacción entre el sistema inmunitario innato y el adquirido3. Hasta el momento ambas respuestas inmunitarias se han caracterizado por separado. En el campo de la inmunología la atención se ha centrado principalmente en el conocimiento de la inmunidad adquirida; sin embargo, el sistema inmunitario natural en mamíferos aún no se ha caracterizado adecuadamente.
La activación del sistema inmunitario innato constituye un paso crucial para el desarrollo de la inmunidad adquirida específica contra antígenos. La respuesta primaria a patógenos en el sistema inmunitario innato está mediada por receptores de reconocimiento de patrones (PRR, de patterns recognition receptors), que reconocen patrones moleculares asociados a patógenos (PAMP, de pathogen- associated molecular patterns), presentes éstos en una amplia variedad de microorganismos4. Entre los PRR figuran de forma destacada los receptores toll-like (TLR, de toll-like receptors), los cuales reconocen con selectividad un amplio número de variados y complejos PAMP, moléculas características de microorganismos como los lipopolisacáridos, las flagelinas, los mananos o los ácidos nucleicos de virus y bacterias. Tras el reconocimiento de estas moléculas propias de microorganismos por parte de los PRR, en especial los TLR, se desencadena una respuesta inmunitaria innata al activar la producción de mediadores inflamatorios como un gran número de interleucinas (IL), los interferones (IFN) y el factor de necrosis tumoral alfa (TNF-α)5.
Funciones y tipos de receptores toll-likeA finales de los años noventa, se identificó el receptor toll como uno de los principales receptores implicados en la defensa (respuesta inmunitaria innata) de la Drosophila frente a las infecciones fúngicas6. Un año después, se demostró que un homólogo mamífero del receptor toll, el receptor toll-like 4 (TLR4), inducía expresión de genes inflamatorios7. Hasta nuestros días se han identificado en mamíferos aproximadamente 15 TLR8,9. En humanos sólo se han descrito 10 TLR funcionales10,11.
Los TLR son proteínas transmembranarias de tipo I que incluyen múltiples copias de LRR (leucine-rich repeats) en el dominio extracelular y un dominio de señalización intracelular compartido por los receptores toll y los receptores de la IL-1 llamado TIR (toll/interleukin-1 receptor)12 (fig. 1). Este dominio13 TIR tiene la habilidad de ligar y activar distintas moléculas, entre ellas la MyD88 (factor 88 de diferenciación mieloide), la adaptadora que contiene el dominio TIR (TIRAP), la adaptadora que contiene el dominio TIR e induce IFN-B (TRIF), la molécula adaptadora relacionada con TRIF (TRAM), cinasas asociadas al receptor de IL-1 (IRAK), factor de necrosis tumoral (TNF), factor 6 asociado al receptor de TNF (TRAF6)14,15; todo ello necesario para activar diferentes vías, tales como las proteincinasas activadas por mitógeno (MAP), los transductores de señales y activadores de la transcripción (STAT) y la vía del factor nuclear κB (NF-κB)10,11.
. El reconocimiento de un ligando —p. ej., lipopolisacárido (LPS)— acciona el TLR, que posteriormente recluta al MyD88 (factor 88 de diferenciación mieloide). El MyD88 interactúa con el TLR a través del TIR (toll/interleukin-1 receptor), que a la vez interactúa con el IRAK (cinasas asociadas al receptor de la interleucina-1). Los factores de transducción de señal, tales como TRAF6 (factor 6 asociado al receptor del factor de necrosis tumoral) estimulados por el IRAK, llevan la señal a través de una serie de fosforilaciones hasta el factor nuclear κB (NF-κB), que finalmente activa la transcripción de genes apropiados para la respuesta inmunitaria.")
Receptor toll-like (TLR). El reconocimiento de un ligando —p. ej., lipopolisacárido (LPS)— acciona el TLR, que posteriormente recluta al MyD88 (factor 88 de diferenciación mieloide). El MyD88 interactúa con el TLR a través del TIR (toll/interleukin-1 receptor), que a la vez interactúa con el IRAK (cinasas asociadas al receptor de la interleucina-1). Los factores de transducción de señal, tales como TRAF6 (factor 6 asociado al receptor del factor de necrosis tumoral) estimulados por el IRAK, llevan la señal a través de una serie de fosforilaciones hasta el factor nuclear κB (NF-κB), que finalmente activa la transcripción de genes apropiados para la respuesta inmunitaria.
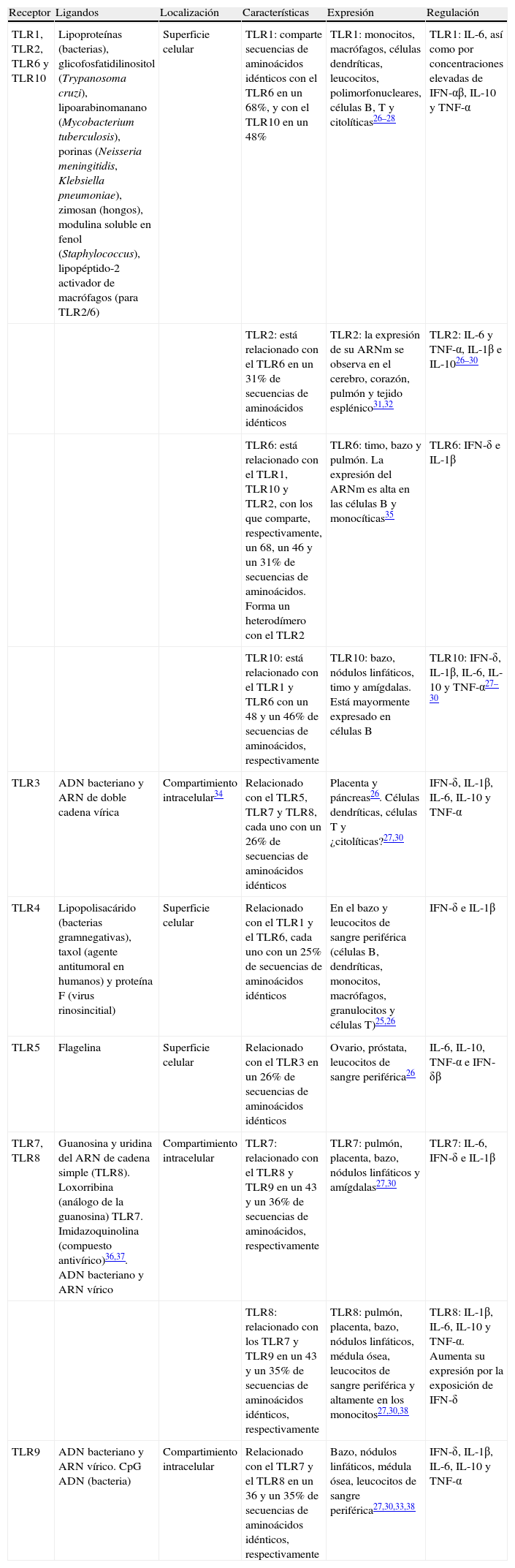
Los TLR1; 2; 4; 5; 6, y 10 se expresan en la superficie celular y migran a los fagosomas (vesícula rodeada de membrana en un fagocito que se forma por la invaginación de la membrana celular y del material fagocitado) tras activarse al reconocer el ligando. Los TLR3; 7; 8, y 9 se expresan en compartimientos intracelulares, principalmente en el endosoma S y el retículo endoplasmático1,4. Cada TLR reconoce un grupo de moléculas características. Los TLR expresados en la membrana celular reconocen moléculas como las lipoproteínas de bacterias grampositivas16 (TLR2 asociado a TLR1 o TLR6), los lipopolisacáridos de las bacterias gramnegativas17 (TLR4) y las flagelinas de los flagelos bacterianos18 (TLR5). Las infecciones por virus ARN, así como por el virus respiratorio sincitial y el de la gripe, son las mayores causas de inflamación de la vía aérea. Los TLR expresados en los compartimientos intracelulares (TLR3; 7; 8, y 9) reconocen ácidos nucleicos de estos virus y de esta forma detectan la infección intracelular19–23 (tabla 1 y fig. 2).
Receptores toll-like (TLR) en humanos
| Receptor | Ligandos | Localización | Características | Expresión | Regulación |
| TLR1, TLR2, TLR6 y TLR10 | Lipoproteínas (bacterias), glicofosfatidilinositol (Trypanosoma cruzi), lipoarabinomanano (Mycobacterium tuberculosis), porinas (Neisseria meningitidis, Klebsiella pneumoniae), zimosan (hongos), modulina soluble en fenol (Staphylococcus), lipopéptido-2 activador de macrófagos (para TLR2/6) | Superficie celular | TLR1: comparte secuencias de aminoácidos idénticos con el TLR6 en un 68%, y con el TLR10 en un 48% | TLR1: monocitos, macrófagos, células dendríticas, leucocitos, polimorfonucleares, células B, T y citolíticas26–28 | TLR1: IL-6, así como por concentraciones elevadas de IFN-αβ, IL-10 y TNF-α |
| TLR2: está relacionado con el TLR6 en un 31% de secuencias de aminoácidos idénticos | TLR2: la expresión de su ARNm se observa en el cerebro, corazón, pulmón y tejido esplénico31,32 | TLR2: IL-6 y TNF-α, IL-1β e IL-1026–30 | |||
| TLR6: está relacionado con el TLR1, TLR10 y TLR2, con los que comparte, respectivamente, un 68, un 46 y un 31% de secuencias de aminoácidos. Forma un heterodímero con el TLR2 | TLR6: timo, bazo y pulmón. La expresión del ARNm es alta en las células B y monocíticas35 | TLR6: IFN-δ e IL-1β | |||
| TLR10: está relacionado con el TLR1 y TLR6 con un 48 y un 46% de secuencias de aminoácidos, respectivamente | TLR10: bazo, nódulos linfáticos, timo y amígdalas. Está mayormente expresado en células B | TLR10: IFN-δ, IL-1β, IL-6, IL-10 y TNF-α27–30 | |||
| TLR3 | ADN bacteriano y ARN de doble cadena vírica | Compartimiento intracelular34 | Relacionado con el TLR5, TLR7 y TLR8, cada uno con un 26% de secuencias de aminoácidos idénticos | Placenta y páncreas26. Células dendríticas, células T y ¿citolíticas?27,30 | IFN-δ, IL-1β, IL-6, IL-10 y TNF-α |
| TLR4 | Lipopolisacárido (bacterias gramnegativas), taxol (agente antitumoral en humanos) y proteína F (virus rinosincitial) | Superficie celular | Relacionado con el TLR1 y el TLR6, cada uno con un 25% de secuencias de aminoácidos idénticos | En el bazo y leucocitos de sangre periférica (células B, dendríticas, monocitos, macrófagos, granulocitos y células T)25,26 | IFN-δ e IL-1β |
| TLR5 | Flagelina | Superficie celular | Relacionado con el TLR3 en un 26% de secuencias de aminoácidos idénticos | Ovario, próstata, leucocitos de sangre periférica26 | IL-6, IL-10, TNF-α e IFN-δβ |
| TLR7, TLR8 | Guanosina y uridina del ARN de cadena simple (TLR8). Loxorribina (análogo de la guanosina) TLR7. Imidazoquinolina (compuesto antivírico)36,37. ADN bacteriano y ARN vírico | Compartimiento intracelular | TLR7: relacionado con el TLR8 y TLR9 en un 43 y un 36% de secuencias de aminoácidos, respectivamente | TLR7: pulmón, placenta, bazo, nódulos linfáticos y amígdalas27,30 | TLR7: IL-6, IFN-δ e IL-1β |
| TLR8: relacionado con los TLR7 y TLR9 en un 43 y un 35% de secuencias de aminoácidos idénticos, respectivamente | TLR8: pulmón, placenta, bazo, nódulos linfáticos, médula ósea, leucocitos de sangre periférica y altamente en los monocitos27,30,38 | TLR8: IL-1β, IL-6, IL-10 y TNF-α. Aumenta su expresión por la exposición de IFN-δ | |||
| TLR9 | ADN bacteriano y ARN vírico. CpG ADN (bacteria) | Compartimiento intracelular | Relacionado con el TLR7 y el TLR8 en un 36 y un 35% de secuencias de aminoácidos idénticos, respectivamente | Bazo, nódulos linfáticos, médula ósea, leucocitos de sangre periférica27,30,33,38 | IFN-δ, IL-1β, IL-6, IL-10 y TNF-α |
ADN: ácido desoxirribonucleico; ARNm: ácido ribonucleico mensajero; CpG: dinucleótido citosina-guanina; IFN: interferón; IL: interleucina; TNF: factor de necrosis tumoral.
 y su expresión. Los TLR1; 2; 4; 5; 6, y 10 se expresan en la membrana celular, y los TLR3; 7; 8, y 9 se expresan en los compartimientos intracelulares.")
El reconocimiento de los ligandos por parte de los TLR, presentes en las células dendríticas y macrófagos, conduce a la rápida producción de citocinas y quimiocinas que indican la presencia del patógeno. Esta respuesta inicia un reclutamiento rápido de células del sistema inmunitario al lugar de la infección y las activa, con lo que se inicia una respuesta inmediata frente al patógeno. Las señales originadas por los TLR promueven la expresión de moléculas de adherencia, tanto en las células epiteliales como en las células hematopoyéticas circulantes.
Entre las células presentadoras de antígeno —aquellas que procesan y presentan el antígeno unido a las moléculas del complejo principal de histocompatibilidad de clase II, figuran los monocitos-macrófagos, las células dendríticas y las células B. Dichas células, en especial las dendríticas, constituyen la interfaz entre ambos tipos de respuestas (innata y adquirida)4. Los ligandos de los TLR provocan que estas células maduren y se conviertan en células presentadoras de antígeno activadas al inducir la expresión de moléculas coestimuladoras (como CD40, CD80 y CD86), necesarias para la activación de los linfocitos T. Muchas IL inducidas por los TLR guían la diferenciación de las células T a linfocitos T helper (CD4+) o linfocitos citotóxicos (CD8+). Los linfocitos T helper-1 (promovidos por la IL-12) producen IL-2, IFN-γ y TNF, y controlan reacciones de inmunidad celular útiles frente infecciones por microorganismos de crecimiento intracelular. Los linfocitos T helper-2 (promovidos por la IL-4) producen IL-4, IL-5 e IL-6, y colaboran en las reacciones de inmunidad humoral, fundamentales para neutralizar toxinas e infecciones por gérmenes de crecimiento extracelular1–3. Existen otros tipos de linfocitos, como los T helper-17 y los T reguladores importantes en el desarrollo y control de la respuesta inmunitaria.
Como ya se ha mencionado, se han identificado 15 tipos de TLR en mamíferos8,9, 13 de ellos, del TLR1 al TLR13, en humanos y ratones. Otras formas equivalentes se han aislado en otras especies de mamíferos; sin embargo, algunos TLR encontrados en humanos no están presentes en otros mamíferos y, por el contrario, otros mamíferos pueden expresar TLR que no se han aislado en humanos. Por ejemplo, los TLR11; 12, y 13 sólo se expresan en ratones. Por el momento sólo se han descrito 10 TLR funcionales en humanos10,11,24,25. Esta circunstancia condiciona o limita el empleo de animales de experimentación como modelos de inmunidad innata extrapolables a la especie humana. Las principales características de los TLR humanos se expresan en la tabla 1.
Los receptores toll-like en el asma y las enfermedades alérgicasLas enfermedades alérgicas como el asma, la rinitis y la dermatitis atópica constituyen un grupo de procesos de elevada prevalencia39–42. Múltiples factores, tanto genéticos como medioambientales, influyen en la susceptibilidad de desarrollarlas41–45. Se dispone de variada y concluyente evidencia que apunta a que la exposición a productos microbianos en la infancia desempeña un papel importante en la maduración posnatal del sistema inmunitario. Se ha hipotetizado que el aumento de la prevalencia, en los países industrializados, de las enfermedades alérgicas en los últimos 20 años podría estar relacionada con la disminución de la carga microbiana en dichas zonas geográficas. Esta hipótesis, conocida como la “hipótesis de la higiene”46,47, se basa en la observación de que en los países desarrollados hay una asociación inversa entre el incremento de las enfermedades alérgicas y la disminución de la exposición microbiana en las primeras fases de la vida, lo cual conduce a un defecto en los mecanismos inmunorreguladores. El descubrimiento de los TLR y de sus acciones proporciona una base inmunológica para estudiar la hipótesis de la higiene46,47.
Todavía hoy quedan numerosas preguntas por responder en relación con la etiología, la patogenia y los fenotipos de asma. Por ejemplo, ¿por qué hay 2 tipos bien diferenciados de asma, alérgica o extrínseca y no alérgica o intrínseca? El asma alérgica se caracteriza por afectar en gran medida a niños y jóvenes, por la presencia de historia personal o familiar de alergia (rinitis, dermatitis atópica), cursa con inmunoglobulina E total y específica elevadas y su mecanismo patogénico se debe en parte a una hipersensibilidad de tipo I (inmediata)48,49. Por el contrario, el asma no alérgica predomina en adultos, rara vez hay historia personal o familiar de alergia y cursa con una inmunoglobulina E en los valores de referencia. Asimismo se plantean interrogantes acerca del papel que desempeñan las diferentes células inflamatorias implicadas en su patogenia. Por ejemplo, se cuestionan el protagonismo universal del eosinófilo en los diferentes fenotipos del asma y su implicación en la clínica real50,51, la falta de respuesta adecuada a la acción antiinflamatoria de los glucocorticoides en algunos pacientes (asma refractaria52), el importante papel que parece desempeñar el neutrófilo en algunos tipos o situaciones de asma, como por ejemplo en el asma ocupacional, las exacerbaciones inducidas por virus53 y el asma de riesgo vital de instauración súbita54. Se considera que dichas células inflamatorias, unidas a la inmunidad innata, desempeñan un papel relevante diferenciado en la patogenia del asma. Esto es algo más que una simple cuestión académica: dada su heterogeneidad inflamatoria, la conocida hoy como enfermedad asmática, más que un proceso homogéneo, podría ser un síndrome, lo que, en consecuencia, podría tener repercusiones de índole práctica por los posibles tratamientos específicos que en el futuro se pudieran administrar.
Por otro lado, la respuesta inmunitaria adquirida en el asma está bien caracterizada e involucra la activación de linfocitos T helper-2 por el alérgeno, con la consecuente inflamación eosinofílica de la vía aérea. De esta forma los eosinófilos activados segregan posteriormente gránulos citotóxicos (proteína principal básica, proteína catiónica eosinofílica), los cuales inducen la hiperrespuesta bronquial y los síntomas consecuentes en el paciente55,56. En el caso del subtipo de asma no eosinofílica (y neutrofílica), donde los síntomas y la hiperrespuesta bronquial persisten a pesar de la ausencia de eosinófilos, el mecanismo patogénico no está bien caracterizado57–66. Sin embargo, hay argumentos para implicar a neutrófilos e IL-857,66 en su patogenia; se trataría de un tipo de respuesta preprogramada del sistema inmunitario natural que se habría conservado a lo largo de la evolución67,68.
Algunos estudios recientes han abierto interesantes expectativas al haber observado que en el asma neutrofílica hay un aumento de la expresión de TLR2, TLR4, CD14 y proteína A del surfactante (SP-A), y que la activación del TLR, por ejemplo por un alérgeno, genera una cascada de señales dirigidas por la activación y translocación nuclear del NF-kB, que da como resultado una respuesta inflamatoria mediada por citocinas (TNF-α, IL-8 e IL-1α)69. Al igual que sucede en el asma alérgica, el asma no alérgica cursa con un incremento de eosinófilos y células T helper-2 en la mucosa bronquial, pero se desarrolla en ausencia de una historia clínica familiar y personal de atopia. Entre los factores etiológicos del asma no eosinofílica, se ha postulado la implicación de alérgenos no identificados, de la autoinmunidad o de infecciones bacterianas o víricas. El asma no alérgica se asocia a menudo con infecciones víricas (Rhinovirus, Coronavirus70) o bacterianas previas, lo cual podría significar que los TLR podrían también estar implicados en esta otra forma clínica de asma66.
El desarrollo de fármacos que actúen sobre los TLR se centra en el uso de ligandos agonistas y antagonistas. Los agonistas son moléculas que se unen a los TLR y generan una respuesta en la célula, mientras que los antagonistas impiden la unión de los ligandos naturales agonistas y, en consecuencia, no provocan ninguna respuesta. En la actualidad se está evaluando el posible papel de diversos agonistas de los TLR como adyuvantes en vacunas, tratamientos antimicrobianos y tratamientos contra la alergia y el cáncer, así como análogos estructurales de los agonistas que se unan al receptor pero no induzcan ninguna señal a la célula. En lo que se refiere al tratamiento del asma y de la rinitis alérgica, la administración de oligodesoxinucleótidos CpG (dinucleótido citosina-guanina), fármaco con capacidad agonista de los TLR, ha resultado eficaz en la prevención y reversión de la inflamación eosinófila bronquial (inducida por antígenos) en modelos de experimentación animal71–73. Los CpG son detectados por los TLR9 presentes en los linfocitos B y en las células dendríticas, cuyo ligando activa múltiples señales en cascada en células que responden como los linfocitos T helper-171.
Los TLR7 y 8 presentes en los compartimientos intracelulares, principalmente en los endosomas, pueden conferir una mayor susceptibilidad a desarrollar enfermedades como el asma, la rinitis y la dermatitis atópica. Recientemente se ha observado que el imiquimod, un nuevo ligando sintético del TLR7 usado con frecuencia en dermatología74, disminuye la inflamación, la hiperrespuesta bronquial, las concentraciones de inmunoglobulina E total en suero y las citocinas en el lavado broncoalveolar, y atenúa la expresión del factor transformador del crecimiento beta en la vía aérea remodelada, en modelos murinos de asma alérgica aguda75.
No obstante, a pesar de los interesantes avances en el conocimiento de las funciones de los TLR y de su posible utilidad en el tratamiento de estas enfermedades inflamatorias, hay que considerar que los expresan numerosas células inflamatorias, entre ellas las células epiteliales, las células T y B, mastocitos y eosinófilos. Esto condiciona la predicción de la respuesta concreta a un agonista o un antagonista de los TLR4. Además, es probable que los productos microbianos que generan esta respuesta contengan diversos ligandos para varios tipos de TLR, con lo que aumentaría aún más la dificultad para predecir una posible respuesta clínica favorable.
Los receptores toll-like en las infeccionesAlrededor de un tercio de la población mundial está infectada por Mycobacterium tuberculosis, pero poco más del 10% de los infectados (inmunocompetentes) desarrollará la enfermedad. Los mecanismos inmunológicos que distinguen cuáles de estas personas presentarán tuberculosis son todavía hoy desconocidos. Se han observado variantes genéticas en los TLR de estos individuos. Entre éstas, polimorfismos del TLR8 aumentan la susceptibilidad a desarrollar tuberculosis pulmonar, y la expresión del TLR8 en los macrófagos está aumentada tras la inoculación de la vacuna de la tuberculosis, la BCG (bacilo Calmette-Guerin)76.
Entre las causas del síndrome de distrés respiratorio agudo se han señalado diversos patógenos, agentes químicos, la gripe aviar H5N1 o el síndrome respiratorio agudo grave. Estudios realizados en un modelo experimental (rata) de síndrome de distrés respiratorio agudo inducido mediante la aspiración de ácido (ALI) y por el virus H5N1 inactivado han demostrado que la mutación del TLR4 confiere una resistencia natural al daño pulmonar agudo. La oxidación fosfolipídica y la producción de citocinas por macrófagos en el pulmón vía TLR4-TRIF fue identificada como causa de ALI. La mutación en ratas del TLR4 demostró una resistencia natural ante el daño pulmonar agudo inducido por ALI, y la señal TLR4-TRIF-TRAF6 es una clave para controlar la gravedad del ALI77. Otros estudios donde se exponía a las ratas a condiciones hiperóxicas atribuyeron al TLR3 un papel relevante en el desarrollo del síndrome de distrés respiratorio agudo, y señalaron que su ausencia durante la hiperoxia confería un efecto protector y que dicho efecto estaba asociado a la inducción de factores proapoptóticos, tales como caspasa-8, caspasa-9, Pten y Bid. Se ha constatado que la vía caspasa-8/Bid tiene un importante papel en la inducción de señales asociadas al daño pulmonar hiperóxido y la muerte celular, tanto in vivo como in vitro. Por tanto, la ausencia genética o la presencia de anticuerpos monoclonales antagonistas de TLR3 parecen atenuar tanto el inicio como la amplificación y favorecer la posterior resolución del daño pulmonar inducido por la hiperoxia. Esto podría hacer pensar en un papel relevante de la inmunidad innata y de los TLR en la patogenia del daño pulmonar agudo78.
En la infección por virus de la gripe se ha evaluado la acción antivírica de agonistas ácido-base nucleicos para la activación de los TLR. Se ha observado que el TLR3 expresado en las células dendríticas, epitelio respiratorio y macrófagos desempeña un papel central en la mediación de la respuesta inflamatoria de la inmunidad innata contra las infecciones víricas. Los virus de la gripe pueden inhibir la capacidad del huésped para producir IFN, así como suprimir los mecanismos de defensa antivíricos del sistema inmunitario. Se ha evidenciado que la administración intranasal en ratones de poli-ICLC (polyriboinosinic-polyribocytidylic acid) y de liposoma encapsulado poli-ICLC, moléculas agonistas de TLR3 inductoras de IFN y linfocitos citolíticos, confiere un alto grado de protección frente al letal virus de la gripe aviar H5N1. La duración de este efecto protector persistió hasta 3 semanas para el liposoma encapsulado poli-ICLC y 2 semanas para el poli-ICLC. De forma similar, el tratamiento previo en ratones con oligonucléotidos CpG (agonista TLR9) confería una completa protección contra la infección del virus de la gripe A79.
Los receptores toll-like en la enfermedad pulmonar obstructiva crónica (EPOC)Para comprender enfermedades como el enfisema es necesario conocer los mecanismos que permiten que el pulmón mantenga su integridad estructural frente a agresiones ambientales constantes como el tabaco80. El enfisema se caracteriza por la pérdida gradual de la elasticidad del pulmón y por la ampliación irreversible del espacio aéreo (insuflación y atrapamiento), generalmente en las últimas décadas de la vida y en relación con la exposición al humo del cigarrillo. Aunque fumar cigarrillos constituye un determinante factor de riesgo asociado al desarrollo de la EPOC, sólo entre el 10 y el 20% de los fumadores importantes la presentan81,82. Esto hace pensar que en el desarrollo del enfisema pulmonar influyen otros condicionantes relacionados con el propio huésped.
En la susceptibilidad a desarrollar enfisema se han implicado muchos genes, concretamente los que regulan la alfa-1-antitripsina83, la enzima macrófago elastasa84, Klotho85, surfactante D86, epóxido hidrolasa microsómica87 y factor nuclear eritroide-2 relacionado con el factor 2 (NrF2)88. Los desajustes entre los procesos oxidativos/antioxidativos o proteasas/antiproteasas podrían ser la causa de las alteraciones observadas en los mencionados genes89. El mecanismo molecular preciso para mantener el equilibrio oxidativo y de proteasas proporcionado es aún desconocido. Estudios en ratas han demostrado que la expresión del TLR4 en las células estructurales del pulmón es necesaria para mantener su arquitectura normal y frenar el estrés oxidativo. En ausencia de TLR4, las células endoteliales expresan concentraciones elevadas de Nox3, oxidantes intracelulares derivados del sistema de la nicotinamida adenindinucleótido fosfato reducido (NADPH) oxidasa, capaces de generar una respuesta inflamatoria que influye en el desarrollo de la EPOC. Por tanto, se cree que el TLR4 actúa como un supresor de la actividad Nox3 endógena en el pulmón, y que su presencia permite mantener la integridad pulmonar mediante la modulación de dicho sistema oxidativo90.
El TLR2 es un receptor que desempeña un papel crucial en la inmunidad innata y en la adquirida. Por este motivo las moléculas con actividad agonista de los TLR2 pueden abrir una nueva estrategia preventiva y terapéutica en las enfermedades alérgicas y en las respiratorias obstructivas crónicas en humanos3,91–93. Como ya se ha mencionado, se ha observado que la expresión de TLR2 y TLR4 sobre los monocitos CD14+ en pacientes con EPOC estable y fumadores sanos disminuye de forma significativa94, por lo que se plantea que quizá la respuesta inmunitaria innata está deprimida en dichos pacientes. La activación de macrófagos alveolares y células alveolares epiteliales vía TLR4-NF-κB, receptor para endotoxinas, expresado y presente en las células epiteliales pulmonares tipo II, también podría desempeñar un papel en la inflamación de la vía aérea de la EPOC mediante la producción y activación de mediadores proinflamatorios tales como IL-895,96. Asimismo, se ha constatado que la exposición aguda al humo del cigarrillo (2 cigarrillos, 2 veces al día durante 3 días) induce inflamación aguda en pulmones de ratas, y ésta es dependiente de la señal TLR4/MyD88 e IL-1R1/MyD8897. También se ha descrito que fumar durante el embarazo atenúa las respuestas inmunitarias mediadas por los TLR, lo que posiblemente hará que aumente en el hijo el riesgo de desarrollar alergias y asma98. También se cree que la expresión en mastocitos humanos y la variación de la codificación del TLR6 podrían desempeñar un papel relevante en la patogenia de la EPOC y del asma99,100.
Los receptores toll-like y otras enfermedades respiratoriasLas células epiteliales respiratorias cumplen un papel importante en los mecanismos de defensa del huésped, así como también en las respuestas inflamatorias. En el tratamiento actual de las enfermedades inflamatorias bronquiales, los glucocorticoides inhalados son habitualmente los fármacos de elección. Reducen de forma efectiva la producción de mediadores inflamatorios, tales como citocinas y quimiocinas, moléculas esenciales para las respuestas de defensa del huésped. El efecto de los glucocorticoides sobre la expresión de los TLR en las células epiteliales respiratorias se ha estudiado recientemente. Se ha observado que la expresión del TLR2 está aumentada por la acción sinérgica derivada de la combinación de TNF-α, IFN-δ y glucocorticoides (dexametasona). Se considera que dicho efecto podría estar relacionado con los receptores de los glucocorticoides, ya que la acción de la dexametasona es abolida por el RU-486, un antagonista de dichos receptores. Esto proporciona a los glucocorticoides otra nueva y beneficiosa función adicional a su bien conocida capacidad antiinflamatoria101.
En la fibrosis quística, la vía aérea afectada representa un entorno potencialmente rico en agonistas TLR. El fenotipo inflamatorio crónico evidente en las células epiteliales de la vía aérea desempeña también un papel importante en las actividades de los TLR. En los pacientes con esta enfermedad se ha constatado un aumento de la expresión del TLR5 significativamente mayor102,103. En la actualidad se está valorando la modulación de los TLR como posible diana terapéutica en el tratamiento de la enfermedad104.
En otro campo de la neumología y no menos importante, estudios en ratones han demostrado que la ventilación mecánica con bajos volúmenes en pulmones sanos induce una respuesta inflamatoria dependiente de los TLR4 (sin alterar la estructura integral del pulmón), aumentando de forma significativa los ligandos endógenos para los TLR4 en el lavado broncoalveolar y la expresión relativa del ARN mensajero de TLR4 y TLR2 en el tejido pulmonar. Esta circunstancia podría abrir expectativas en la mejora del conocimiento de los posibles cambios en la respuesta inmunitaria que experimentan los pacientes ventilados105.
En definitiva, múltiples estudios experimentales coinciden en otorgar a los TLR un papel importante en los mecanismos de defensa e inmunorregulación. Hay evidencias que apuntan a qué posibles alteraciones en dichos receptores podrían intervenir en la patogenia inflamatoria de diversas enfermedades respiratorias, particularmente el asma, la EPOC y las infecciones. Un previsible mayor conocimiento de las moléculas que los potencian (agonistas) o inhiben (antagonistas) podría incrementar en un futuro el arsenal terapéutico frente a las mencionadas enfermedades. En la actualidad se están efectuando diversos ensayos clínicos que, en diferentes fases de desarrollo, están explorando su eficacia y seguridad. Confiemos en que en el futuro se confirmen estas buenas expectativas y podamos disponer de este nuevo tratamiento farmacológico.







