


Amyloidosis is a systemic disease caused by abnormal deposition of amyloid material that is detected with Congo red staining and is difficult to diagnose. Involvement of the tracheobronchial tree is rare and is a challenge for pulmonologists because of the wide differential diagnosis of this disease. We present two cases where tracheobronchial affectation has been observed: in one of them as a primary disease, and in another as secondary affectation. The use of bronchoscopic techniques is essential for the diagnosis of tracheobronchial involvement. In the absence of an effective drug therapy, local management of this disease with endoscopic techniques for bronchial repermeabilization is able to provide clinical improvement and expand the treatment options and prognosis in this disease.
La amiloidosis es una enfermedad sistémica producida por el depósito anómalo de material amiloide; tiene la peculiaridad de detectarse con la tinción rojo Congo y es de difícil diagnóstico. La afectación del árbol traqueobronquial es muy poco frecuente y constituye un reto para el neumólogo debido al amplio diagnóstico diferencial de esta enfermedad. Se presentan 2 casos en los que se ha objetivado la afectación traqueobronquial: en uno de ellos como enfermedad primaria y en otro como afectación secundaria. El uso de técnicas broncoscópicas es primordial para el diagnóstico de la afectación traqueobronquial. En ausencia de un tratamiento médico eficaz, el manejo local de esta enfermedad con técnicas endoscópicas de repermeabilización bronquial consigue una mejoría clínica y amplía las opciones terapéuticas y pronósticas en esta enfermedad.
Amyloidosis is a systemic disease of unknown origin which is characterized by extracellular deposits of proteins with a specific structural conformation that gives them a characteristic apple-green birefringence when stained with Congo red. They are deposited mainly in the heart, kidneys and liver. Diseases caused by abnormal amyloid deposits can be classified into primary or secondary, local or systemic, based on their distribution. Therefore, pulmonary involvement by amyloidosis can be both secondary and primary, systemic or localized. It has an estimated incidence of between 5 and 10 cases per million persons/year. However, primary pulmonary involvement is much rarer,1 and presents a challenge for the pulmonologist, as the use of bronchoscopy techniques is essential for diagnosis and to provide optimal treatment.
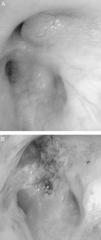
CasesCase 1A 46-year-old male who attended our clinic due to unproductive cough, moderate effort dyspnea of recent onset and temperature of 38°C. His only history was that he smoked 45 packs/year, with no symptoms of chronic bronchitis. On physical examination, he had baseline oxygen saturation measured by pulse oximetry (SpO2) of 93% on room air, and rhonchi in both hemithorax on pulmonary auscultation. Chest X-ray revealed alveolar consolidation in the right base and incipient consolidation in the left apex, and a nodular image on the apical segment of the left lower lobe. Blood tests showed leukocytosis of 17.6×109/L with neutrophilia 86%, and C-reactive protein 19mg/dL. Due to suspected pulmonary neoplasia, a chest computed tomography (CT) scan was performed, showing ground glass opacities in the left upper lobe and posterior basal segments of both lower lobes; it also showed multiple nodular (centroacinar) and tree-in-bud images, localized in the apical segment of the left lower lobe and in posterior basal segments with a tendency to cluster, with no nodal formations. Fibrobronchoscopy was performed, revealing an obstruction of >80% of the lumen of bronchi B1-B6D due to infiltration of the submucosa and nodular lesions (Fig. 1A), as well as irregular stenosis of the right basal bronchi. He also had carinal lymph node enlargement and tracheal stenosis in the upper third. The biopsy showed respiratory epithelium with deposits of amyloid material; immunohistochemical techniques with Congo red confirmed the finding as amyloidosis. An extension study was carried out, discarding systemic disease, so the diagnosis of localized primary tracheobronchial amyloidosis was established.
Case 2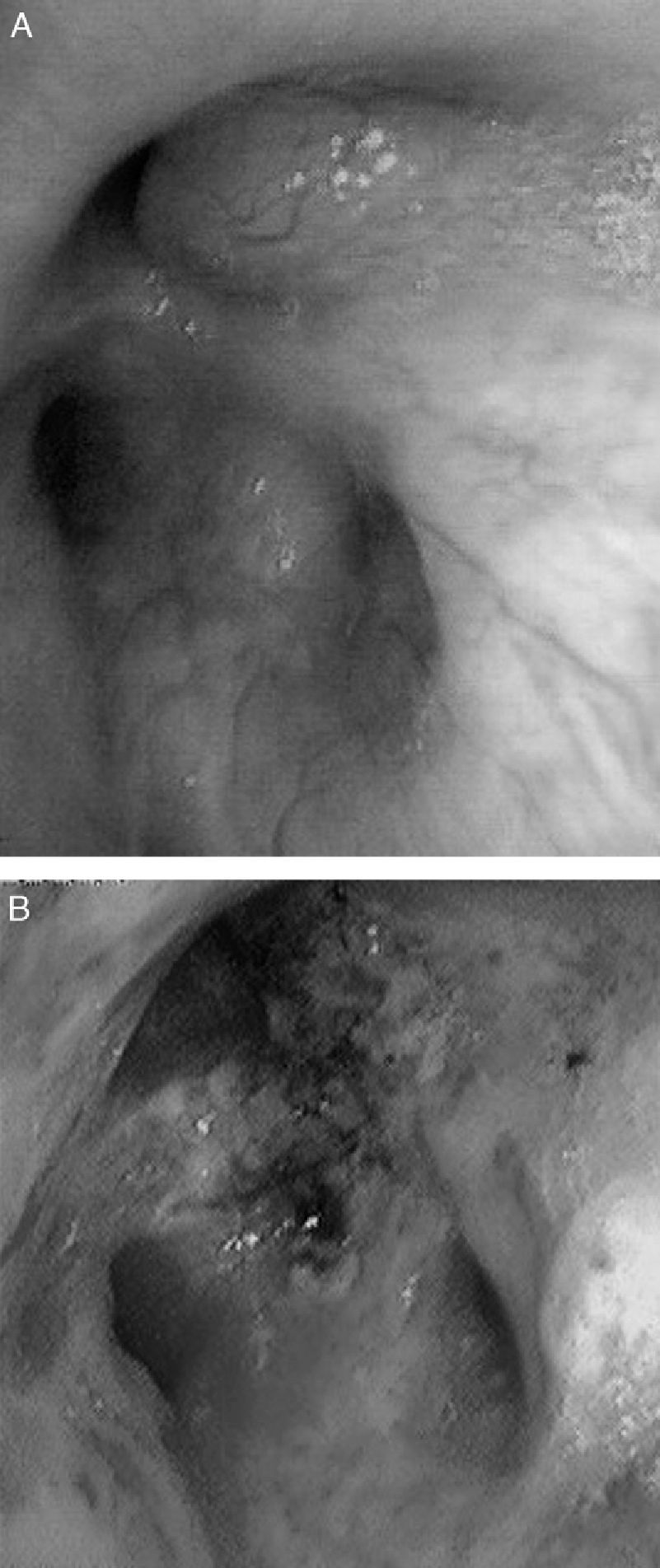
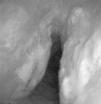
57-Year-old woman with repeated episodes of hemoptysis with no other symptoms. She had a history of hypertension for which she was on treatment with valsartan, chronic nasal obstruction and no toxic habits. The physical examination was normal, as were additional tests performed: laboratory tests and chest X-ray. A bronchoscopy was carried out, detecting multiple friable, bleeding nodules on the anterior and posterior side of the trachea, one of which was ulcerated (Fig. 2). The histopathology study demonstrated the presence of eosinophilic amorphous material that exhibited birefringence with polarized light, corresponding to amyloid material. An extension study of the disease was carried out, in which amyloid material was also detected in the subcutaneous fat and bone marrow biopsy. The patient was diagnosed with primary systemic amyloidosis with lung and bone marrow involvement.
Discussion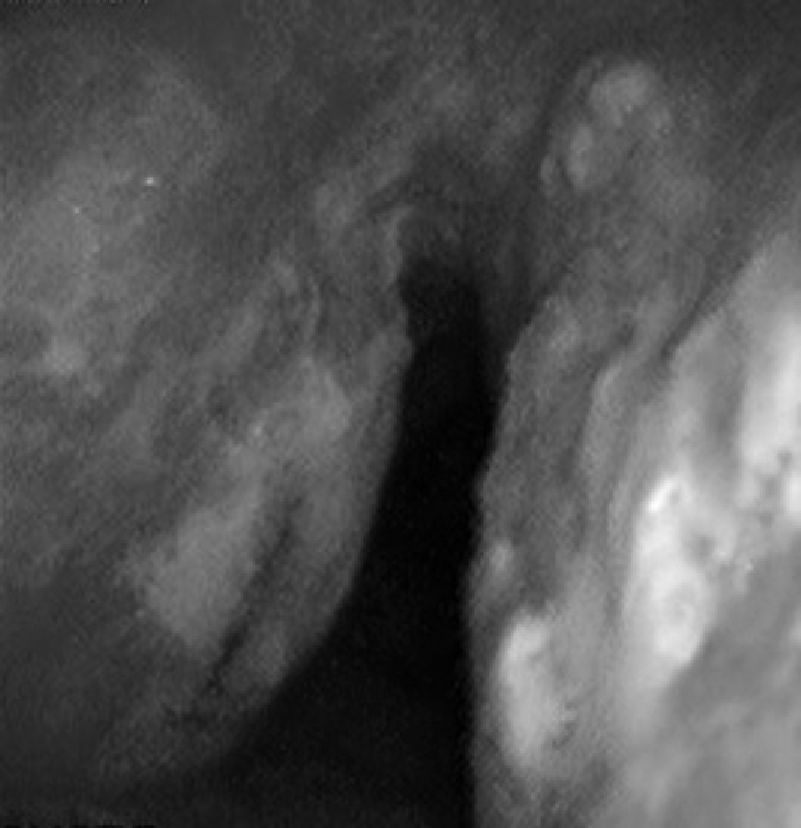
Amyloidosis is a rare disease of unknown etiology characterized by abnormal deposition of fibrillar proteins that typically exhibit apple-green birefringence under polarized light after Congo red staining. Involvement is variable, and there may be primary, secondary or senile disease. It may be systemic or well localized in an organ or tissue.2 Pulmonary involvement by amyloid is exceptional, whether it forms part of a systemic disease or is localized. Five forms of primary pulmonary amyloidosis have been described: tracheobronchial, nodular (solitary or multiple), senile pulmonary, mediastinal-hilar and diffuse interstitial.3 Tracheobronchial amyloidosis is the most common presentation,3 and is characterized by amyloid deposits in the tracheobronchial tree only.
Amyloidosis affects mainly males (2:1) and in middle age (50–60 years). The clinical presentation, in the case of systemic disease, varies depending on the organs involved. In the case of pulmonary involvement, cough (74%), audible wheezing (70%), dyspnea (60%), hemoptysis (50%) and stridor (30%) are more common.4 More than half of cases may present as obstructive pneumonia, bronchiectases or atelectases,4 so its identification poses a major challenge, even forming part of the differential diagnoses of various conditions such as asthma, recurrent pneumonias and neoplasia.
Several imaging techniques have been used to diagnose the disease, although the most useful is high resolution CT, which enables the form of pulmonary involvement to be distinguished.5 Much newer is the use of PET-CT, which shows intense uptake of 18-fluorodeoxyglucose (18-FDG) by the amyloid material. It is used in order to assess activity, and is useful in patient follow-up.6
Definitive diagnosis is made through histopathological study of the affected area, making it essential to perform bronchial endoscopic techniques when the airways are affected. Several bronchoscopic patterns have been suggested: nodular, pseudotumor, polypoidal and sub-mucosal,7 which together with endobronchial ultrasound (EBUS) (to discard mediastinal involvement) results in a full assessment of the disease by the bronchoscopist. Systemic involvement can be assessed by echocardiography and urinary protein measurements, which evaluate the cardiac and renal involvement, respectively, as these are the two organs most often affected.
No effective medical treatments have been described for pulmonary amyloidosis, although there are therapeutic bronchoscopy techniques which are effective for the management of this disease, with the primary aim of repermeabilizing the airways. To that end, Nd:YAG laser has been used to treat bronchial obstructions due to amyloid deposits,8 while symptomatic improvement has also been achieved in the mid-term with the use of cryotherapy.9 The rigid bronchoscope enables both endoscopic resection of an amyloid obstruction and subsequent placement of a Dumon endobronchial stent to maintain the airway permeable for longer.10 External beam radiation therapy (EBRT) achieves significant clinical improvement,11–13 although it requires individualization of treatment, given the possible complications due to the radiation.11
Colchicine treatment has classically been used when there is systemic involvement. Better survival has subsequently been demonstrated with dexamethasone as monotherapy,14 and especially with the combination of prednisone and melphalan.15 Autologous stem cell transplant is performed where possible.2 When the involvement is localized in the tracheobronchial tree or lung parenchyma, treatment with systemic drugs is not indicated.2
With respect to follow-up of these patients, the highly experienced University of Boston working group suggests serial pulmonary function tests and CT imaging as the best way of assessing the status of the airways and disease progression.16
The prognosis is related to systemic involvement, with greater mortality in patients with cardiac involvement; its main cause of death is sudden death.17 In the case of localized pulmonary involvement, the prognosis improves; the main causes of death in these patients are respiratory failure and massive hemoptysis.
The patient in case 1 was treated on two occasions with Nd:YAG laser to repermeate the airways (Fig. 1B), owing to the stenosis that he presented due to amyloid infiltration, achieving clinical and functional improvement. With respect to the patient in case 2, she received treatment with a combination of prednisone and melphalan, with a good response and partial resolution of the tracheal lesions, with no functional or symptomatic impairment.
FundingNone.
Conflict of InterestThe authors declare that there is no conflict of interest.
Please cite this article as: Berraondo J, et al. Manejo de la amiloidosis traqueobronquial mediante técnicas broncoscópicas terapéuticas. Arch Bronconeumol. 2013;49:207–9.







