



The pulse oximeter, a non-invasive method of measuring oxygen transported by hemoglobin inside the blood vessels, is one of the most widely used devices in medical practice. It emits light with 2 wavelengths of 660 nm (red) and 940 nm (infrared) that are characteristic of oxyhemoglobin and reduced hemoglobin, respectively, and calculates the percentage of oxyhemoglobins by comparing the absorption of light during the pulsatile wave with the baseline level.1 Correlation between oxygen saturation and the partial pressure of oxygen in arterial blood is determined by the oxyhemoglobin dissociation curve.
In clinical practice, a level of oxygen saturation well below the expected limits2 means that many etiologies must be ruled out, some of which are quite rare. We report a case of hemoglobinopathy with altered oxygen affinity detected by pulse oximetry.
Our patient was a 53-year-old man, farmer by profession, and former smoker who had started smoking at the age of 13, and had quit 10 years previously; pack-year index: 15; allergic to penicillins and intolerant to tramadol; congenital ventricular septal defect, with no current impact on health. Family disease history included mother with chronic heart disease and father who died of non-Hodgkin’s lymphoma. He has a healthy sister.
He was referred to our clinic from primary care for a 6-month history of dyspnea on intense exertion, and no other respiratory symptoms.
Physical examination showed no relevant findings, no cyanosis, no tachypnea, no other signs of chronic respiratory failure, but basal oxygen saturation was 76%–77%, surprisingly low and surprisingly well tolerated, confirmed by different devices. Blood gas analysis was carried out in which partial pressure of oxygen in arterial blood was below normal, confirming hypoxemia with oxygen saturation of 76%, with a partial pressure of oxygen in arterial blood of 41 mmHg. Chest radiograph was normal. Spirometry was also performed, and values were normal in all cases (FVC: 5580–117%, FEV1: 4470–123%, FEV1/FVC: 80%, MEF: 25–75%: 3340–100%).
To rule out the most common causes of hypoxemia, we performed a computed tomography of the chest which showed only a persistent left superior vena cava, and hypoplastic brachiocephalic trunk, thus excluding, among other problems, thromboembolic disease at a radiological level. Diffusion testing was also performed, which was normal.
Given the patient’s cardiological history, we requested a specific reassessment to rule out any kind of anteriovenous shunt: transthoracic and transesophageal echocardiograms and a bubble study were performed, ruling out any active ventricular septal defect. Cardiac catheterization was also performed, showing normal left ventricular ejection fraction, and ruling out changes in coronary artery lumens and in the pulmonary and systemic circulation.
Clinical laboratory tests were performed, including full blood count, biochemistry, kidney function, liver profile, thyroid markers, etc.), showing no changes.
Finally, after contacting the hematology department, we performed hemoglobin electrophoresis: the patient had hemoglobin with abnormal electrophoretic mobility of up to 35%. P50 was also calculated, which in our patient was 34.5, consistent with hemoglobinopathy with altered oxygen affinity. Given this suspicion, we requested the Hospital San Carlos in Madrid to identify the anomalous hemoglobin. After DNA sequencing, the patient was determined to have hemoglobin Rothschild.
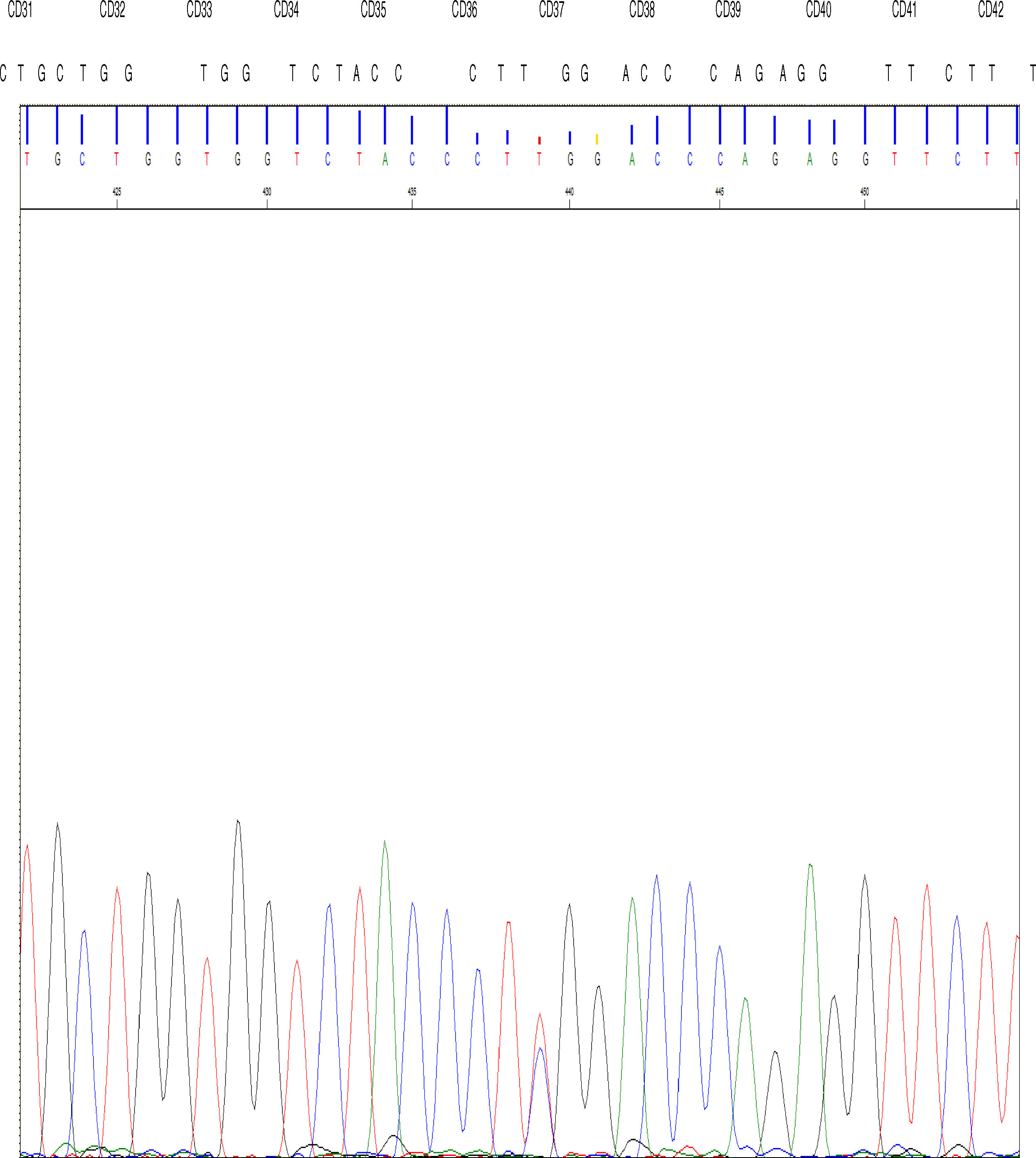
The DNA study identified the TGG > CGG mutation at codon 37 in the 2nd exon of the gene β in the allele that determines the replacement of amino acid tryptophan by arginine at position 3 of the C helix of the chain β encoded by the mutated allele.
High performance liquid chromatography was also performed, which showed abnormal hemoglobin in the fraction corresponding to hemoglobin D; after new sampling for arterial (p50 value of 34.5) and venous blood gases, we concluded that this was hemoglobin Rothschild [β37 (C3) Asp > Tyr; HBB: c.112 T > C] (Fig. 1).
Hemoglobin consists of a tetramer of polypeptide chains of globins, a pair of α chains, 141 amino acids in length, and a pair of β chains of 146 amino acids.3 The main hemoglobin in adults is HbA, which has an α2β2 structure. Each heme fraction can bind to a single molecule of oxygen, so that each molecule can carry up to 4 molecules of oxygen. The key properties that are altered in hemoglobin diseases are solubility and the reversible union of oxygen, which depends particularly on hydrophilic amino acids, hydrophobic amino acids that line the heme pocket, a histidine in the F helix, and the amino acids that form the contact surfaces of α1β1 and α1β2. The alteration that modifies oxygen affinity is located in these regions.3
Hemoglobin Rothschild is a mutation of the hemoglobinβ chain, where it substitutes the tryptophan residue at position β37(C3) with arginine. Tryptophan in B37 (C3) is a contact point at the α1β2 and α2β1 interfaces, and the replacement of tryptophan by arginine leads to destabilization of the interfaces affected during oxygenation and deoxygenation: a deviation from normal oxygen levels is expected, but this deviation occurs only at the molecular level and has no apparent clinical manifestation in patients, who usually remain asymptomatic.4,5 It should be noted that the partial pressure at which the hemoglobin is half saturated is p50, HbA has a p50 of 25 ± 2 mmHg, whereas hemoglobin Rothschild [β 37(C3) RTP → Erg] has a higher p50 due to its low affinity to oxygen.4–6
Altered oxygen saturation in our patient, determined by pulse oximeter, was the initial reason for performing this study. After identifying the cause of the problem to be hemoglobinopathy and explaining to the patient the limited clinical implications of this finding, he was able to minimize his problems and symptoms, which had been exaggerated by his knowledge of the abnormality and his fear of possible disease. We believe that, in patients presenting low oxygen saturations with no obvious cause and little clinical concordance, this type of hemoglobinopathy should be taken into consideration because despite its rarity, it can appear in routine practice.
In addition, to complete the family study and to identify other cases, pulse oximetry was used to identify our patient's sister and 2 nephews as carriers.
When a patient presents with low oxygen saturations with no apparent pathological cause to explain the hypoxia,4,5,7 we must bear in mind an oxygen affinity alteration7–9 of hemoglobin, since many of these conditions develop without symptoms5 and uncommon diseases can be detected by measuring saturation using a pulse oximeter,10,11 as in the case which we report here.
Please cite this article as: González García LM, et al. Una causa infrecuente de saturación arterial de oxígeno baja: hemoglobinopatía de Rothschild. Arch Bronconeumol. 2019;55:657–659.







