



Carbocysteine can reduce the frequency of acute exacerbations and improve respiratory symptoms to a certain extent in severe to very severe chronic obstructive pulmonary disease (COPD) patients. The objective of the study was to evaluate the efficacy of carbocysteine on the rate of exacerbations and pulmonary function for mild-to-moderate COPD patients.
MethodsIn this phase 4, multicenter, double-blind, randomized, placebo-controlled, parallel-group trial, we randomly assigned mild-to-moderate COPD patients in a 2:1 ratio to treatment with carbocysteine (500mg, thrice daily) or matched placebo for 12 months. Eligible participants were 40–80 years of age. The coprimary outcomes were the annual rate of exacerbations of COPD (mild, moderate or severe) during the 12-month trial period or the difference in the FEV1 before bronchodilator use at 12-month from baseline.
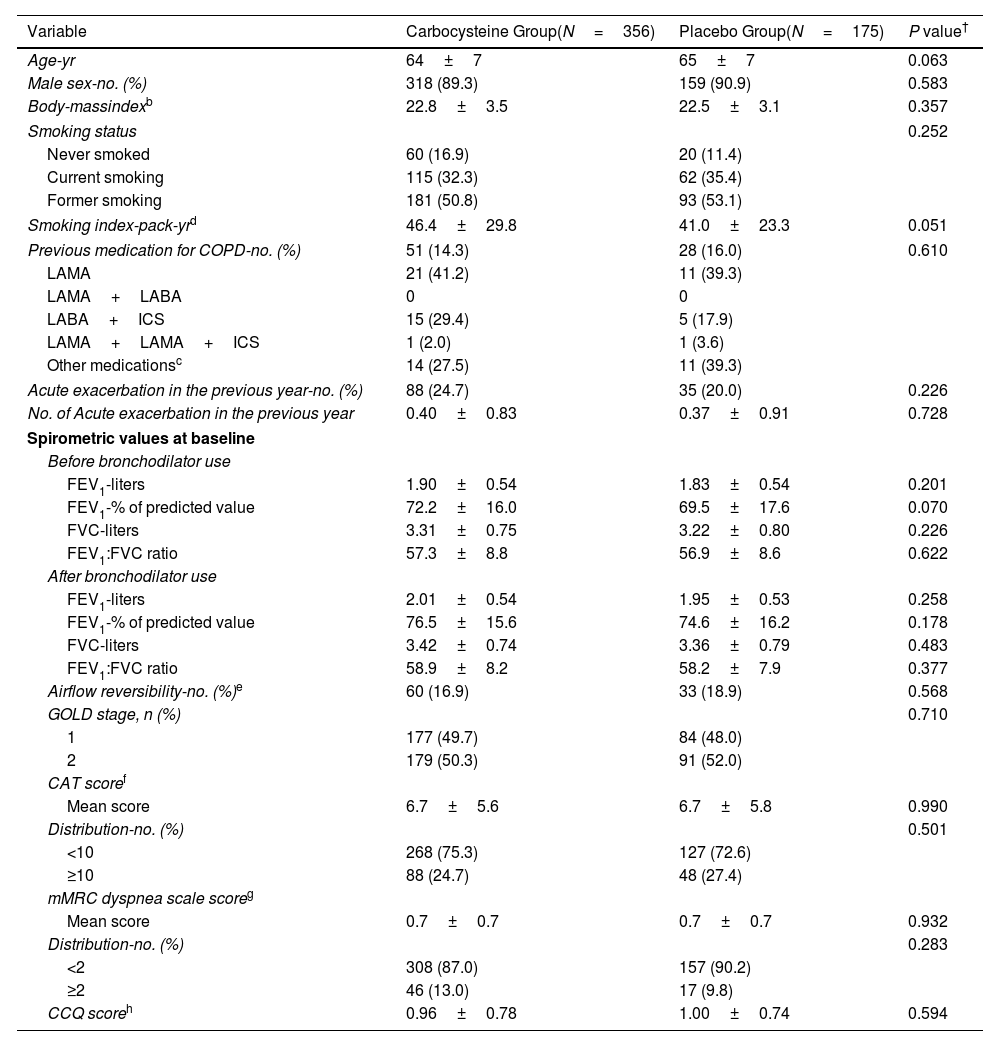
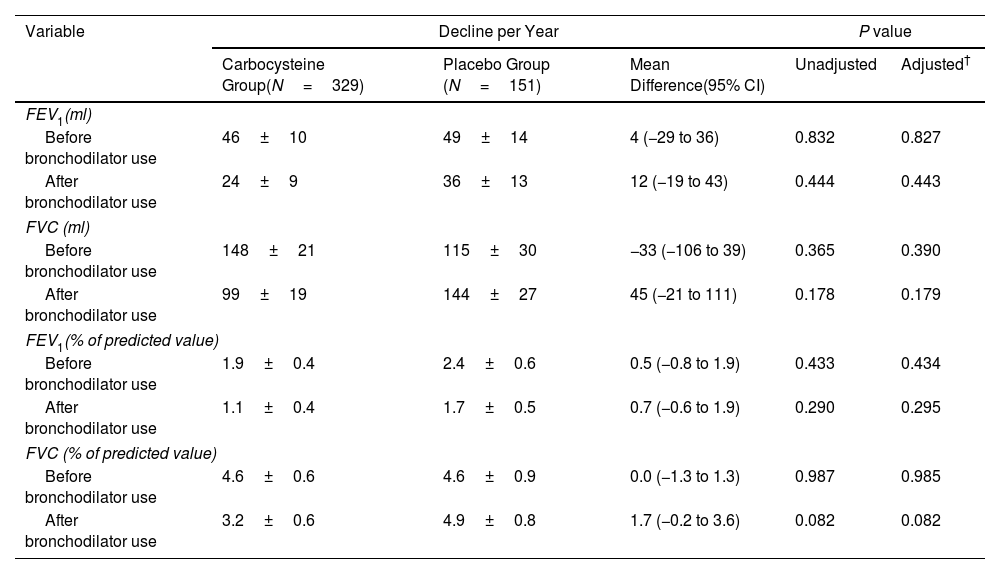
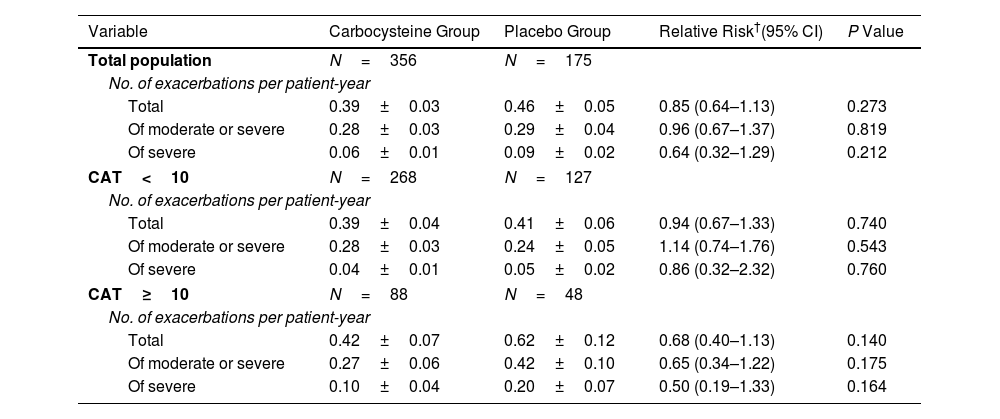
ResultsOwing to slower-than-anticipated recruitment caused by the COVID-19 pandemic, recruitment of an estimated sample size of 732 patients stopped after 539 patients. The sample size was indeed reached for the annual rate of exacerbations but not for pulmonary function. Among 539 patients, 362 were randomized to receive carbocysteine and 177 to receive matched placebo. There was no significant difference in the annual rate of exacerbations of COPD between carbocysteine group and placebo group (0.39 vs. 0.46 per patient year; relative risk [RR], 0.85; 95% confidence interval [CI], 0.64–1.13; P=0.273). Based on the available sample size, the difference in the change of FEV1 before bronchodilator use at 12 months between carbocysteine group and placebo group has not been observed (46±12 vs. 50±17ml; mean difference, 6 ml; 95% CI, −24 to 36; adjusted P=0.700).
ConclusionsOur findings suggested that carbocysteine might not significantly reduce the annual rate of total exacerbations in patients with mild-to-moderate COPD. The findings may have been compromised by an overestimation of the efficacy of carbocysteine on reducing exacerbations in mild-to-moderate COPD and potential confounding by baseline imbalances. The efficacy on lung function could not be adequately evaluated.
Clinical trial registered with Chictr.org.cn (ChiCTR1800016712).








