
Pulmonary neuroendocrine tumors (PNT) encompass a broad spectrum of tumors including typical carcinoid (TC) and atypical (AC) tumors, large-cell neuroendocrine carcinoma (LCNEC) and small-cell lung cancer (SCLC). Although no variety can be considered benign, AC and TC have a much lower metastatic potential, are usually diagnosed in early stages, and most are candidates for surgical treatment. Several chemotherapy (CT) regimens are available in the case of recurrence or in advanced stages, although scientific evidence is insufficient. LCNEC, which is currently classified alongside large-cell carcinomas, have molecular features, biological behavior and CT sensitivity profile closely resembling SCLC. Pathological diagnosis is often difficult, despite the availability of immunohistochemical techniques, and surgical specimens may be necessary. The diagnostic tests used are similar to those used in other lung tumors, with some differences in the optimal tracer in positron emission tomography. The new TNM classification is useful for staging these tumors. Carcinoid syndrome, very rare in PNT, may cause symptoms that are difficult to control and requires special therapy with somatostatin analogs and other drugs. Overall, with the exception of SCLC, new trials are needed to provide a response to the many questions arising with regard to the best treatment in each lineage and each stage.
Los tumores neuroendocrinos pulmonares (TNP) abarcan un amplio espectro de tumores que incluyen los carcinoides típicos (CT) y atípicos (CA), el carcinoma neuroendocrino de células grandes (CNCG) y el carcinoma microcítico de pulmón (CMP). Aunque ninguna variedad puede considerarse benigna, los CA y CT tienen un potencial metastásico mucho menor, habitualmente se diagnostican en estadios tempranos y la mayoría son subsidiarios de tratamiento quirúrgico. Se dispone de varias pautas de quimioterapia (QT) en caso de recidiva o en estadios avanzados, aunque la evidencia científica es insuficiente. Los CNCG, encuadrados en la clasificación actual junto a los carcinomas de células grandes, tienen rasgos moleculares, conducta biológica y perfil de sensibilidad a la QT que los asemejan más a los CMP. Con frecuencia su diagnóstico anatomopatológico es difícil, pese al uso de técnicas de inmunohistoquímica, y pueden ser necesarias muestras quirúrgicas. Las pruebas diagnósticas a utilizar son similares a las empleadas en otros tumores de pulmón, con algunas diferencias en cuanto al trazador óptimo que se usa en la tomografía de emisión de positrones. La nueva clasificación TNM es de utilidad en la estadificación de estos tumores. El síndrome carcinoide, muy infrecuente en los TNP, puede dar síntomas de difícil control que requieren medicación especial con análogos de la somatostatina y otros fármacos. En general, y con la excepción del CMP, se necesitan nuevos ensayos que den respuesta a numerosos interrogantes sobre el mejor tratamiento a aplicar en cada estirpe y cada estadio.
Neuroendocrine cells are derived from primitive pluripotent cells and are characterized by production of neurotransmitters and lack of axons and synapses. The tumors they produce in various organs are classified into 3 groups, anterior, middle and posterior, according to their embryonic origin in the gastrointestinal tract. These tumors may arise not only from the lung but from various regions of the digestive system.1,2 Pulmonary neuroendocrine tumors (PNT) represent approximately 25%–30% of primitive lung cancers (LC). Of these, 80% are anaplastic small cell lung cancers (SCLC), 12% are large cell neuroendocrine carcinomas (LCNEC) and the remaining 8% are typical carcinoid (TC) and atypical carcinoid (AC) tumors, the latter being the most uncommon.3–5 As discussed below, the prognosis for these tumors is very variable and although some, such as TCs, are associated with good life expectancy, they should not be described as “benign” as they all have metastatic potential.3 At present, these tumors are classified according to their grade of malignancy as: low grade (TC), intermediate grade (AC) and high grade (LCNEC and SCLC) (Table 1). The latest World Health Organization lung tumor classification (2004) also includes other entities, such as diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) and tumorlets.6–8 The latter are accumulations of neuroendocrine cells generally found in the airways that invade the basement membrane. Their appearance is similar to that of carcinoid tumors but they measure less than 0.5cm. They are usually found by chance and do not cause symptoms.7 Although the significance and natural history of DIPNECHs has not been well determined, they are considered by many as the earliest manifestation of neuroendocrine disease and classed as precancerous lesions.8 They are often associated with chronic bronchopulmonary lesions, such as bronchiectasis and fibrosis, so in this setting they are considered reactive lesions. They rarely occur in previously normal lungs, and if they do, they may develop into carcinoid tumors.3
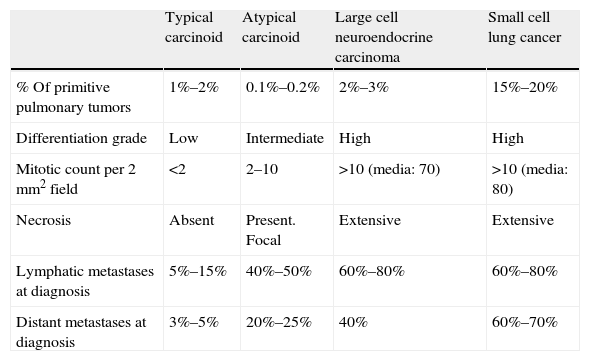
Characteristics of Pulmonary Neuroendocrine Tumors.
| Typical carcinoid | Atypical carcinoid | Large cell neuroendocrine carcinoma | Small cell lung cancer | |
| % Of primitive pulmonary tumors | 1%–2% | 0.1%–0.2% | 2%–3% | 15%–20% |
| Differentiation grade | Low | Intermediate | High | High |
| Mitotic count per 2mm2 field | <2 | 2–10 | >10 (media: 70) | >10 (media: 80) |
| Necrosis | Absent | Present. Focal | Extensive | Extensive |
| Lymphatic metastases at diagnosis | 5%–15% | 40%–50% | 60%–80% | 60%–80% |
| Distant metastases at diagnosis | 3%–5% | 20%–25% | 40% | 60%–70% |
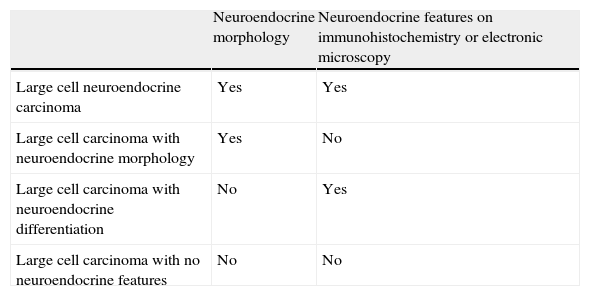
With regard to the histological classification of these tumors, some criteria for the definition of the various types are shown in Table 1. Surgical specimens often have to be examined to determine diagnosis, particularly in the case of LCNEC. When only small biopsy specimens from fiberoptic bronchoscopy are available, it may be very difficult to establish a precise diagnosis. In addition to the mitotic index, the presence or absence of necrosis and the structure of the tumor, the immunohistochemical detection of various neuroendocrine markers, such as chromogranin A, synaptophysin or CD56 and other molecular changes may be an aid to identification.3,7,9 LCNEC does not always display all the features that characterize this lineage; therefore, in terms of the overall group of large cell carcinomas, the possible spectrum of neuroendocrine differentiation varies widely6,7 (Table 2). Moreover, the Ki-67 proliferation marker has recently been identified as possibly useful for distinguishing between low and high grade tumors, particularly when only small biopsies or cytological specimens are available.3,8 Despite the availability of these techniques, differential diagnosis of the disease is often complicated and careful evaluation by experienced pathologists is required.8 Recent studies suggest that as LCNEC is relatively rare and difficult to diagnose, it may be underdiagnosed, and although it frequently resembles conventional non-small cell lung cancer (NSCLC) both clinically and morphologically, its biological characteristics and prognosis are different, and more importantly, its sensitivity to various chemotherapy (CT) regimens appears to be closer to that of SCLC.9
Morphologic Features and Neuroendocrine Differentiation in Large Cell Lung Carcinomas.
| Neuroendocrine morphology | Neuroendocrine features on immunohistochemistry or electronic microscopy | |
| Large cell neuroendocrine carcinoma | Yes | Yes |
| Large cell carcinoma with neuroendocrine morphology | Yes | No |
| Large cell carcinoma with neuroendocrine differentiation | No | Yes |
| Large cell carcinoma with no neuroendocrine features | No | No |
Low grade PNTs appear at an earlier age (mean 40–50 years) than most LCs and are not clearly associated with tobacco smoking or gender, while high grade PNTs are mainly diagnosed in men over 60 years of age and are closely related with smoking. Carcinoid tumors are central in 75% of the cases and initial symptoms are cough, hemoptysis, wheezing, recurrent pneumonia or chest pain.2,5 Paraneoplastic syndromes are uncommon: carcinoid syndrome, considered characteristic, is more common in the gastrointestinal presentation of the disease and only appears in 1%–3% of tumors of pulmonary origin. Others, including Cushing's syndrome (1%–2% of cases), acromegaly, etc., are even less common. However, when a thorough endocrine evaluation is performed, hypersecretion may be found in up to 15% of patients, the majority of whom do not have clinical symptoms.5
Peripheral pulmonary carcinoid tumors are usually detected by chance. The biological behavior of LCNEC and SCLC is more aggressive, so they are more likely to produce metastasis and are often advanced when diagnosed. SCLC commonly occurs with paraneoplastic syndromes of various types (endocrine, neuromuscular, hematological, etc.), the most common of which is insufficient antidiuretic hormone secretion that may occur in up to 40% of cases.10
Diagnosis and StagingLike other more common LCs, the starting point for suspected diagnosis is generally an abnormal image on chest X-ray. Other imaging tests to be performed for defining the anatomical characteristics of the tumor and possible distant metastases, including chest computed tomography (CT), magnetic resonance, scintigraphy, etc., are also similar, so the guidelines for the diagnosis and staging of LC in general11–13 are also applicable in these patients. Since most tumors are central, a biopsy specimen is usually obtained by fiberoptic bronchoscopy. If the tumor is peripheral, transthoracic biopsy or aspiration may be the first option, although the small size of cytological specimens and biopsies can be a limiting factor for precise diagnosis, as has been mentioned previously. Thus, after the specimen has been examined, a larger specimen may have to be obtained in a second intervention.
For staging, carcinoid tumors show some specific biological features that limit the efficacy of tests such as positron emission tomography (PET). These slow-growing tumors have low glucose uptake, so 18F-fluordeoxyglucose PET is of little use, particularly in TCs.2 However, new radiotracers, such as 111In-octreotide or 68Ga-DOTATATE, have been developed that, given their special affinity for the somatostatin receptors often present in low grade tumors, are of great utility in diagnosis and staging.5 The usefulness of both 18F-fluordeoxyglucose and 68Ga-DOTATATE was recently evaluated with PET/CT in a group of neuroendocrine tumors of different grades. The authors found an inverse infinity for both radiotracers for low and high grade tumors. High grade tumors showed great avidity for 18F-fluordeoxyglucose and low avidity for 68Ga-DOTATATE, while the opposite was true in low grade tumors.1468Ga-DOTATATE is not yet available in many centers, but it is currently thought to be of great value in planning interventions and for detecting recurrence of low grade carcinoid tumors.
As discussed below, the most common therapeutic option for low grade PNTs is surgery, but the rapid dissemination of high grade tumors often rules out this approach. However, classification using the tumor, node, metastasis (TNM) system is useful for both groups. The latest edition of the TNM classification is not only more accurate, but has also shown greater universality and its prognostic value in both SCLC and carcinoid tumors has been confirmed.15–18
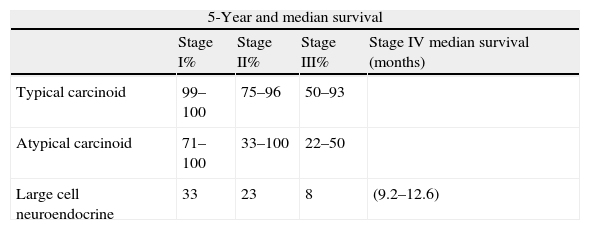
PrognosisAs mentioned above, the PNT spectrum encompasses a range of tumors with important pathological, biological and clinical differences. Consequently, life expectancy also differs greatly. Most carcinoid tumors are diagnosed in TNM stages I and II, are mostly operable, and 5-year survival is over 90%. In contrast, most LCNECs and SCLCs are diagnosed in advanced stages, are candidates only for treatment with CT and radiotherapy, and long-term survival is very low (Table 3).2,9,19
Pulmonary Neuroendocrine Tumor Survival.
| 5-Year and median survival | ||||
| Stage I% | Stage II% | Stage III% | Stage IV median survival (months) | |
| Typical carcinoid | 99–100 | 75–96 | 50–93 | |
| Atypical carcinoid | 71–100 | 33–100 | 22–50 | |
| Large cell neuroendocrine | 33 | 23 | 8 | (9.2–12.6) |
| Limited disease % median (months) | Extended disease % median (months) | |
| Small cell lung cancer | 20–25 (18–24) | <10%a (10) |
The Spanish Neuroendocrine Tumor Group (EMETNE) analyzed survival data from a large series of resected carcinoid tumors, finding that the histological subtype, the presence or absence of mediastinal node involvement and distant metastases were the factors that most influenced prognosis.20,21 Other authors studying the AC subgroup found that a high mitotic index, size of primary tumor >3.5cm and female sex were independent indicators for poor prognosis.22
Given the difficulties encountered in the pathological diagnosis of LCNECs and their rarity, few studies analyzing prognostic factors have been published, although TNM staging appears to be of value in this respect.5,9 Although survival among resected patients varies widely depending on staging, it seems that, if staging is similar, patients with LCNEC have poorer life expectancy that those with NSCLC.9 Lastly, in the case of SCLCs, many studies on prognostic indicators have been available for some time.23 In this review, only the expediency of using the new TNM classification in this entity is mentioned, although the simple and traditional subdivision into two groups, limited or extended disease (limited to one hemithorax or extended beyond that), still has practical value.
TreatmentIf SCLCs are excluded, PNTs represent a small proportion of primitive lung cancers and the various types show wide biological heterogeneity. This explains the insufficient number of clinical trials comparing large, homogeneous patient groups, and the consequent low grade of scientific evidence for many of the treatment regimens discussed. With the exception of SCLCs, there are few randomized studies supporting the currently used regimens. This does not mean that there are no firm indications backed up by experience, such as resection of most TNM stage I and II carcinoid tumors, given the excellent survival results of this procedure. The treatment of each of the various PNT groups is discussed individually below.
Carcinoid TumorsTCs and ACs, of low and intermediate grade, respectively, are almost always candidates for surgery, and most TNM stage I and II tumors have high life expectancy (Table 3).2,5,20,21 Lobectomy is the most commonly performed procedure, although for central tumors, sleeve resections may be performed to preserve lung parenchyma.2,19 In elderly patients with poor lung function and peripheral tumors, smaller resections, such as segmentectomy or wedge resection may be the only option, although these procedures are not considered adequate from an oncological point of view, given the small but real metastatic potential of these tumors.19 The relapse rate ranges between 5% and 30% and is higher in ACs and patients with mediastinal node involvement.2,19,20 For this reason, adjuvant treatment with CT and/or thoracic radiation therapy (TRT) is recommended, although efficacy has not been demonstrated.2,5,19,24 There are no clear, evidence-based recommendations on the best approach in the case of incomplete resection, distant relapse or even in those rare cases in which the carcinoid tumor at the time of diagnosis is in a sufficiently advanced stage (TNM IIIB or IV) as to be considered inoperable. A recent guideline recommends TRT for TCs with mediastinal involvement and combined CT and TRT for ACs,25 although the grade of evidence is low. The most commonly used CT regimen is based on platinum derivatives and etoposide (similar to the standard SCLC regimen), although limited experiences have also been published with other drugs, such as streptozotocin, doxorubicin with 5-fluorouracil, temozolamide or everolimus.5,25,26 The rate of favorable responses (mainly partial remissions) did not exceed 20%–30% in the best series.4,5,19,26
In the symptomatic treatment of carcinoid syndrome, somatostatin analogs, such as octreotide and similar derivatives, e.g., lanreotide, have been used for their antisecretory effects. They are administered intramuscularly and have shown moderate efficacy in controlling symptoms, although they do not lack adverse effects.1,2,5 In patients with symptomatic advanced gastrointestinal neuroendocrine tumors, a recent randomized trial showed better results with the combination of octreotide and everolimus (an inhibitor of the metabolic pathways involved in the pathogenesis of these tumors, e.g., m-TOR) than with octreotide alone.27 Interferon-alpha28 and combinations of interferon with the antiangiogenic bevacizumab29 have also been studied. However, the very limited experience available due to the rarity of carcinoid syndrome and the fact that these compounds are not free of adverse effects suggest that patients who are potential candidates for these treatments should be referred to specialist centers.5
Large Cell Neuroendocrine CarcinomaAs discussed above, LCNEC shares some morphologic features with other lineages and is often difficult to identify, particularly when the available biopsy specimens are small. Although LCNEC is classified within the group of non-microcytic large cell carcinomas, it is more similar in its molecular profile and biological behavior to SCLC.9,30 In any case, for TNM stages I and II, surgery is considered standard treatment.2,5,31 However, poor long term survival (between 27% and 67% at 5 years in stage I resected patients31) suggests that multimodal adjuvant treatment with CT and/or TRT may be appropriate. Various retrospective analyses have found greater survival in patients receiving adjuvant CT with platinum derivatives and etoposide, similar to the standard regiment for SCLC.9,31 These results appear to be confirmed by a prospective Japanese study which reported a reduced rate of relapse and longer 5-year survival in patients who received adjuvant treatment.31
Since LCNEC is generally diagnosed in advanced stages, CT is the only treatment option. Given the diagnostic difficulties of typing and confusion about positioning this entity in the histologic classification of LCs, some recent treatment guidelines do not appear to distinguish between this variety and other SCLCs.25 However, several retrospective analyses coincide in finding improved survival and response rates with CT regimens similar to those used for SCLC (cisplatin and etoposide) compared to those used for NSCLS.9,32–34 A recent phase II trial found a partial remission rate of 46.7% with combined irinotecan and cisplatin, very similar to the 50%–60% reported in previous studies.32–34 These data suggest that LCNECs are sensitive to the same agents used in SCLC, although the response rate in the former is somewhat lower. Lastly, although the rate of epidermal growth factor receptor mutations in these tumors is unknown, the report of a case of LCNEC with positive mutation and an excellent response to gefitinib36 suggests that this aspect may be of therapeutic interest.35
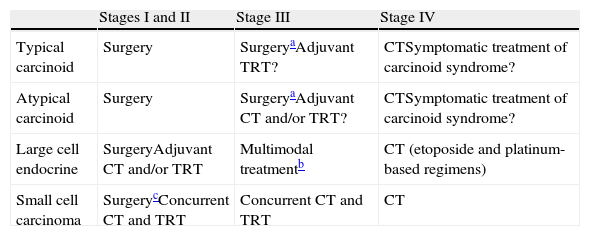
A simplified schematic of treatment options is presented in Table 4.
Treatment of Pulmonary Neuroendocrine Tumors.
| Stages I and II | Stage III | Stage IV | |
| Typical carcinoid | Surgery | SurgeryaAdjuvant TRT? | CTSymptomatic treatment of carcinoid syndrome? |
| Atypical carcinoid | Surgery | SurgeryaAdjuvant CT and/or TRT? | CTSymptomatic treatment of carcinoid syndrome? |
| Large cell endocrine | SurgeryAdjuvant CT and/or TRT | Multimodal treatmentb | CT (etoposide and platinum-based regimens) |
| Small cell carcinoma | SurgerycConcurrent CT and TRT | Concurrent CT and TRT | CT |
For the much more common SCLC, numerous treatment guidelines based on large randomized trials are available.25,37 Surgery is currently reserved for a small number of stage I patients with an acceptable general condition, after possible hidden metastases have been ruled out by thorough evaluation (less than 3% of all SCLCs).37 In other patients with disease limited to the chest (LD), the treatment of choice is based on the concurrent combination of CT and TRT (Table 4). Early initiation of hyperfractionated radiation therapy twice a day from the first or second cycle along with platinum-based CT plus etoposide every 21 days for a total of 4–6 cycles is recommended. For extended disease (contralateral and/or extrathoracic pulmonary metastases) (ED), CT consisting of etoposide plus platinum or irinotecan should be administered. Prophylactic cranial irradiation is currently indicated in the case of objective, complete or partial response, in both LD and ED,25,37 given the high rate of central nervous system relapse. Lastly, with regard to new targeted or specific anti-target treatments, none of the compounds tested has shown efficacy in this type of LC.37
Since PNTs, with the exception of SCLCs, make up a tiny proportion of all LCs, the subtypes have very distinct biological features and histologic typing is often difficult, it is understandable that the available scientific evidence is insufficient to serve as a basis for therapeutic recommendations. To achieve this objective, as already proposed in the IASLC meetings,9 international collaboration among the various existing registries is essential to allow clinical trials with sufficient numbers of patients to be conducted.
FundingThis paper did not receive funding from any source.
Conflict of InterestsThere are no conflicts of interests associated with this paper.
Please cite this article as: Sánchez de Cos Escuín J. Diagnóstico y tratamiento de los tumores pulmonares neuroendocrinos. Arch Bronconeumol. 2014;50:392–396.







