



Cystic fibrosis is a single gene, autosomal recessive disorder, in which more than 1900 mutations grouped into 6 classes have been described. It is an example a disease that could be well placed to benefit from personalized medicine. There are currently 2 very different approaches that aim to correct the basic defect: gene therapy, aimed at correcting the genetic alteration, and therapy aimed at correcting the defect in the CFTR protein. The latter is beginning to show promising results, with several molecules under development. Ataluren (PTC124) is a molecule designed to make the ribosomes become less sensitive to the premature stop codons responsible for class i mutations. Lumacaftor (VX-809) is a CFTR corrector directed at class ii mutations, among which Phe508del is the most frequent, with encouraging results. Ivacaftor (VX-770) is a potentiator, the only one marketed to date, which has shown good efficacy for the class iii mutation Gly551Asp in children over the age of 6 and adults. These drugs, or a combination of them, are currently undergoing various clinical trials for other less common genetic mutations. In the last 5 years, CFTR has been designated as a therapeutic target. Ivacaftor is the first drug to treat the basic defect in cystic fibrosis, but only provides a response in a small number of patients. New drugs capable of restoring the CFTR protein damaged by the most common mutations are required.
La fibrosis quística es una enfermedad autosómica recesiva monogénica, de la que se han descrito ya más de 1.900 mutaciones agrupadas en 6 clases y que constituye un ejemplo de lo que podría ser una enfermedad bien situada para poder beneficiarse de la medicina personalizada. En la actualidad, 2 enfoques muy diferentes tienen por objetivo corregir el defecto básico: la terapia génica, dirigida a corregir la alteración genética, y la terapia encaminada a corregir el defecto a nivel de la proteína CFTR. Esta última comienza a dar resultados prometedores con diversas moléculas en desarrollo. Ataluren (PTC124) es una molécula diseñada para que los ribosomas se vuelvan menos sensibles a los codones de parada prematuros responsables de las mutaciones clase i. Lumacaftor (VX-809) es un fármaco corrector que está dirigido a mutaciones de clase ii, entre las que figura la más frecuente (Phe508del), con prometedores resultados. Ivacaftor (VX-770) es un fármaco potenciador, el único comercializado hasta el momento, que ha demostrado una buena eficacia para la mutación de clase iii, Gly551Asp, en niños mayores de 6 años y adultos. Además, diversos ensayos están probando estos fármacos o la combinación de ellos para otras mutaciones genéticas menos frecuentes. En los últimos 5 años, la CFTR ha sido designado como una diana terapéutica. Ivacaftor es el primer fármaco que trata el defecto básico de la fibrosis quística, pero solo da respuesta a un escaso porcentaje de los pacientes. Se precisan nuevos fármacos capaces de restaurar la proteína CFTR causada por mutaciones más comunes.
The completion of the human genome project was a relevant milestone for medical knowledge, providing the information necessary for understanding the unique characteristics of each individual.1 The logical consequence of this knowledge would be to be able to apply specific diagnostic tests and treatments to each patient based on their individual genetic information. This new form of medical care is called personalized medicine.2 However, despite the great progress that has led to the knowledge of the human genome, its translation into diagnostic and personalized treatments has been less than expected. At present, steps are being in this direction with two major initiatives: systems biology3 and pharmacogenetics.4 The ultimate goal of these initiatives is to develop a medical practice customized to the characteristics of each individual that can predict the onset or course of a particular disease, allow appropriate prevention strategies to be established and, finally, enable the patient to take part in the decision making. This has been called P4 medicine.5
Cystic fibrosis (CF) remains the most common and lethal genetic disease among Caucasians. It occurs at a rate of 1 in 2500–6000 newborns, depending on the region and ethnic origin, and in a proportion of healthy carriers, varying between 1:20 and 37.6 In Spain, thanks to the progressive introduction of neonatal screening programs in the different communities, a lower incidence of CF is now being seen than was previously estimated in 2009, i.e., 1/4430 live births in Galicia, 1/4339 in Castile-Leon, 1/5376 in Murcia, 1/5840 in Catalonia, and 1/6602 in the Balearic Islands.7 It is estimated that there are 70000 CF sufferers worldwide.8 The disease is caused by mutations in the gene encoding the regulatory protein cystic fibrosis transmembrane conductance regulator (CFTR), a chloride channel involved in the release of adenosine triphosphate and regulation of other ion transport channels. This protein is expressed in respiratory epithelial cells, pancreas, biliary tract, sweat glands and genitourinary system. Its alteration leads to an abnormality in ion transport, so that patients produce thick, sticky mucus that clogs the ducts of the organ where it is located and so the alteration presents multisystemic effects that determine the wide range of clinical manifestations of CF. Despite major advances in the treatment of CF that have resulted in longer survival (present median is estimated in 37.5 years),9 there is still a long way to go to ensure that patients with CF have a quantity and quality of life similar to that of subjects without the disease. In this context, new treatments to decrease morbidity and increase survival are necessary.
CF is an example of a disease well-positioned to take advantage of personalized medicine. On one hand, it is a monogenic disease, caused by the mutations in a specific gene. The pathophysiology of the entity is well characterized and the therapeutic targets are clear. Furthermore, diagnosis of the disease requires genetic testing for the identification of the disease type, so the exact genetic defect is determined in each case.10
At present, two very different approaches are targeted at correcting the basic defect: gene therapy, aimed at correcting the genetic alteration, and molecule therapy, aimed at correcting the functional defect at the protein level. The focus of gene therapy is the introduction of normal gene copies in the airways of CF patients. It involves the insertion of a recombinant viral vector, the DNA of which has been extracted and replaced by the new therapeutic DNA. The viral vector serves as a vehicle for inserting the new DNA into the target cell. Various types of viruses, such as adenovirus or lentivirus, have been used to date. Furthermore, non-viral particles, such as nanoparticles capable of inserting DNA, have also been developed.11 However, the results so far have been poor, because the duration of expression of the introduced gene was short with both types of vectors.12 The UK Gene Therapy Consortium is developing a phase II clinical trial to evaluate the clinical effectiveness of an optimized plasmidic/liposomal DNA vector.13 The recruitment target is 130 patients and results are expected in 2014 (NCT01621867).14
On the other hand, therapy directed at restoring the function of the CFTR protein has been more successful. In recent years, results are beginning to come through on drugs that act directly on the CFTR protein. In fact, in January 2012, the first drug for correcting Gly551Asp mutation defects was marketed in the USA. In the following sections we will review the available information on the progress of personalized medicine for CF and available treatments aimed at correcting the defect causing the disease at the protein level. In this review, the nomenclature used for the description of CFTR gene mutations developed by the Human Genome Variation Society15 will be used.
Mutations and Protein DefectCF is an autosomal recessive hereditary disease, so the mutation must be present in both copies of the CFTR gene to be affected. To date, over 1900 CFTR gene mutations associated with the disease have been identified in the coding sequence, messenger RNA or other elements. Mutations in the CFTR gene are available for consultation in the Cystic Fibrosis Mutation Database.16 The first mutation described, and the most common worldwide, is Phe508del, but there are other specific mutations with varying frequency among different ethnic groups. In Spain, the average frequency of Phe508del mutation is between 50% and 60% of all of the studied chromosomes, the second most frequent is Gly542X with 4%–8%, followed by Asn1303Lys in 2%–4% of cases. The mutations described to date are classified into six types or classes according to the mechanism causing the disease.17 These types of mutations are summarized in Fig. 1. Class I mutations lead to a premature stop codon in the messenger RNA which prevents translation of the complete protein. Thus, the protein produced is short and non-functioning. Class II mutations encode a structurally abnormal and misfolded protein that is removed by the endoplasmic reticulum before reaching the cell surface. The most common mutation in CF, Phe508del, belongs to this group. In the case of mutations in classes III to VI, proteins reach the cell surface, but do not function properly. Class III mutations cause a decreased channel activation, so channels remain closed. Class IV mutations cause a decrease in ion conductance through the channel. Class V mutations encode minor proteins resulting in a reduced amount of CFTR in the cell surface, so that a certain function occurs, but at a reduced level. Finally, class VI mutations lead to a shortened half-life due to protein instability and can also damage the regulation of neighboring CFTR channels in the cell surface.
CFTR Modulators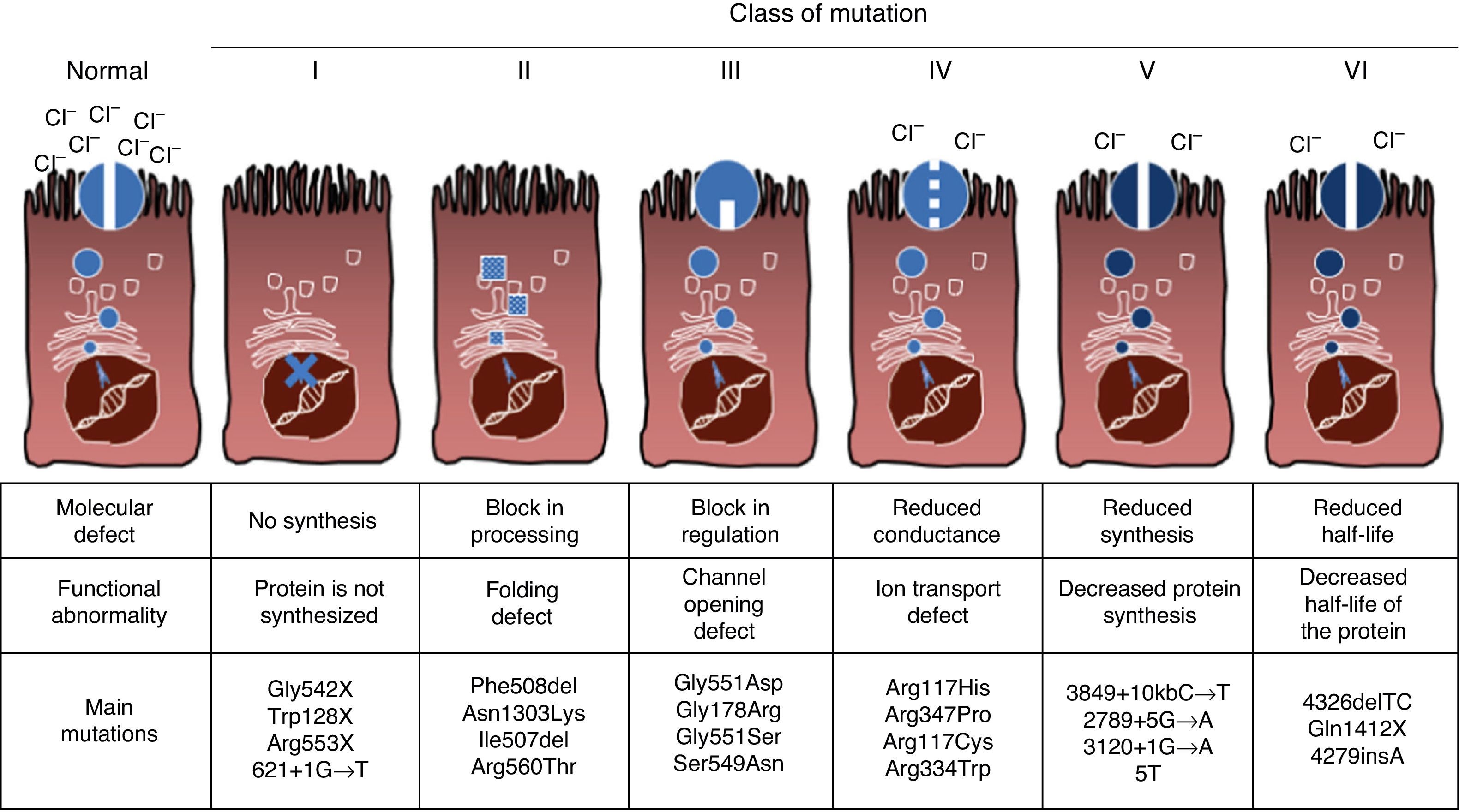
Three main classes have been identified in the development of drugs for repairing CFTR protein.11 The first group are premature stop codon suppressors (class I mutations). These drugs prevent identification of this premature stop codon, so that protein synthesis can continue until completion. The second group are the CFTR correctors. These compounds are designed to correct defects in the transport of folded protein (class II mutations) to the cell membrane, where it may be able to function almost normally. The third group consists of the so-called CFTR potentiators. These are drugs designed to target the CFTR protein on the cell surface in order to improve its function. Thus, these potentiators can act on class III, IV, V and VI mutations. Currently, numerous molecules using these different mechanisms are under investigation, one of which has already reached the market: Ivacaftor (VX-770) is a CFTR potentiator approved in the USA in January 2012 for the treatment of CF patients older than 6 years of age who have the Gly551Asp mutation.
Class I Mutation TreatmentsApproximately 10% of CF cases are caused by class I mutations. The first drugs used for this class were aminoglycosides. Several years ago, gentamicin was reported to have the ability to mask the premature stop codon preventing synthesis of the CFTR protein. This is achieved by insertion of an amino acid that enables the ribosome to continue reading the gene, producing a full-length protein. Preclinical studies demonstrated that the protein could be synthesized in a mouse model and 35% of protein function was recovered in vitro.18,19 The effect of the intravenous administration of gentamicin was assessed in two studies of CF patients with various types of class I mutations. One was conducted in the USA in five patients,20 and another included 18 patients in France.21 However, although the response was positive, the results varied widely, so the benefit was not universal. Moreover, toxicity issues with aminoglycosides contributed to an unfavorable profile.
A synthetic alternative is ataluren (PTC124; PTC Therapeutics, South Plainfield, NJ, USA). This is a molecule designed to enable ribosomes to read the genetic information while “skipping” the premature stop codon, producing a functional CFTR protein.22 The pharmacokinetics of ataluren have been demonstrated in animal models and in phase II trials. In two small preliminary studies,23,24 a group of CF patients treated with oral ataluren showed improvement of the electrophysiological abnormalities of the disease, with an increase in the number of cells in the nose expressing the protein on their surface. Subsequently, another small study of 19 patients evaluated different doses of ataluren administered orally every 8h, with improvements in CFTR activity and clinical parameters and a good safety profile.25 The results of a phase III clinical trial of ataluren that had not yet been formally published were reported in the 2012 North American Cystic Fibrosis Conference. A total of 238 patients older than 6 years were randomized to receive ataluren 10, 10, 20mg/kg or placebo every 8h for 48 weeks. There were no significant differences in FEV1 in ataluren versus placebo after 48 weeks of treatment (−2.5% ataluren vs −5.5% placebo, P=ns). When stratified by chronic nebulized antibiotic use, there was a 6.7% difference in mean change after 48 weeks in favor of ataluren for patients not on tobramycin, while there was no difference in change in FEV1 in those receiving nebulized tobramycin. Something similar occurred with the percentage of exacerbations, where the difference between both groups was significant. When stratified, the patients in the ataluren group not on nebulized tobramycin showed a percentage decrease in exacerbations of 43% compared to the placebo group. Nasal potential difference and sweat chloride tests showed no difference between groups, irrespective of the use of nebulized antibiotics. The authors concluded that the benefits were greater in patients not receiving chronic antibiotic therapy with nebulized aminoglycoside, speculating that tobramycin and ataluren interacted at a ribosomal level, producing antagonism when used simultaneously.26
Class II Mutation TreatmentsClass II mutations, being the most frequent mutation of the disease (Phe508del), are present in a large number of CF patients, making them a primary target in CF research. Various molecules have been studied, most of which have been developed by Vertex Pharmaceuticals Inc. The first corrector compound, lumacaftor (VX-809), showed good in vitro efficacy, improving chloride transport in 14%.27 However, the results were somewhat disappointing in patients, because the improvements in chloride concentration in sweat was very small (7mmol/l) and there were no changes in nasal potential.28
The effects of the CFTR potentiator ivacaftor (VX-770), a drug for class III mutations (see description below), have also been investigated in patients who were homozygous for Phe508del. The results of the DISCOVER study show that ivacaftor is not associated with a significant improvement in FEV1, quality of life or the number of exacerbations versus placebo.29
Since lumacaftor can assist the delivery of Phe508del-CFTR to the cell surface and ivacaftor increases opening time and chloride conduction through the epithelial cell, improvement of the underlying Phe508del defect may be possible with the combination of both molecules. In vitro studies of combined ivacaftor and lumacaftor in respiratory epithelia with Phe508del mutation have shown that lumacaftor increases CFTR chloride transport by 15% on its own, and when ivacaftor is added, transport increases to almost 30%. This combination of drugs has been investigated in a phase II study in patients with Phe508del mutation. Full results have not been yet published, but initial data suggest a beneficial effect on lung function in homozygous Phe508del, but not in heterozygosity.30 Two clinical trials in patients 12 years or older homozygous for the mutation Phe508del are being developed to evaluate combined ivacaftor and lumacaftor; these are the TRAFFIC (NCT01807923)31 and TRANSPORT (NCT01807949)32 studies. The results might allow about half of the CF population to receive CFTR modulating therapy. However, studies are needed to evaluate the effect of combination therapy in Phe508del heterozygous individuals.
Another alternative is the corrector compound VX-661, and studies are currently ongoing. Its effectiveness is being tested alone and in combination with ivacaftor (VX-770) and the results will soon be available (NCT01531673).33
Class III Mutation TreatmentsIvacaftor (VX-770) is a CFTR potentiator that modulates the function of the abnormal protein.34 This molecule was originally designed to enhance CFTR function in cultures of respiratory epithelium cells carrying a single Gly551Asp mutation.34 It is the first drug approved in the USA and Europe for the treatment of CF in patients carrying the Gly551Asp mutation. After the capacity of ivacaftor to improve chloride transport through the cell membrane was demonstrated in vitro, the first phase II trials were conducted with 39 patients.35 In this study, CFTR protein function improved three days after starting treatment, reaching chloride concentrations in sweat of up to normal levels. With these results, two further clinical studies followed: STRIVE study, in 144 patients aged 12 years or more36 and ENVISION study including 52 children between 6 and 11 years of age.37 Both studies included patients with at least one Gly551Asp mutation and FEV1 between 40% and 105% that were initially followed for a period of 14 days and then randomized to receive 150mg of oral ivacaftor or placebo twice a day for a period of 48 weeks. After completing 48 weeks of treatment, the patients were given the opportunity to continue in a longitudinal open-label study, the PERSIST study (NCT01117012),38 for 96 weeks.
In STRIVE, patients in the ivacaftor group had an improvement of 10.6% in FEV1 (primary endpoint) starting from day 15 of treatment, which was maintained during the 48-week study. Furthermore, a decrease in the chloride concentration in sweat was observed (mean: −48.7mmol/L), with improved quality of life, a 55% reduction in exacerbations and a weight gain of 2.7kg.
The results of the ENVISION study coincide largely with those of the study in adolescents and adults, with the difference that quality of life did not reach statistical difference. The adverse effects more frequently observed in the treatment group in both STRIVE and ENVISION studies were upper respiratory tract infections, nasal congestion, sore throat, dizziness and skin rash. The preliminary results after the first 12 weeks of the PERSIST study reveal that improvements in lung function (FEV1), respiratory symptoms, and weight gain among patients treated with ivacaftor are maintained during this period. Moreover, the subgroup of patients who switched from placebo to ivacaftor at baseline in PERSIST experienced a 10.8% improvement in FEV1 at 15 days, and 13% at 12 weeks, together with a reduction in exacerbations.39
Despite the good results with ivacaftor in the treatment of the Gly551Asp mutation in children older than 6 years and adults at 48 weeks, some issues remain to be resolved. Firstly, the drug has not been tested in children under 6 years. However, it seems sensible to correct the defect before irreversible damage occurs, considering that lung involvement begins before the age of six. A trial in this age range is currently ongoing (NCT01705145).40 Secondly, another alternative would be to also try other class III mutations. In this regard, in vitro studies on nine other mutations have shown very similar results,41 so it is reasonable to expect similar results in patients. There is an ongoing phase III clinical trial in patients older than 6 years with other class III mutations (KONTINUE and KONNECTION studies; NCT01614470).42,43 Thirdly, although no treatment is available for the rest of the mutation classes (IV–VI), CFTR potentiators may be equally beneficial in these cases. A clinical trial is being conducted in this respect, evaluating the efficacy of ivacaftor in the Arg117His class IV mutation (KONDUCT study; NCT01614457).44 Finally, effectiveness and long-term safety of ivacaftor beyond 48 weeks have not yet been established. The G551D Observational Study (GOAL; NCT01521338)45 is an observational study following patients older than 6 years receiving ivacaftor. It is aimed at reporting the efficacy and safety of long-term ivacaftor, along with other outcomes of interest that include inflammatory mediators in sputum, mucociliary clearance, and gastrointestinal pH. Results are expected in late 2013.
ConclusionsCF is an example of a disease well-positioned to take advantage of personalized medicine. At present, two very different approaches aim to correct the basic defect: gene therapy aimed at correcting the genetic alteration, and therapy with molecules aimed at correcting the functional defect at the protein level. The latter is beginning to show promising results for various molecules in development, and one of them (ivacaftor) is already marketed for Gly551Asp class III mutation, with excellent results in children older than 6 years, adolescents and adults. The final objective is to provide corrector and potentiator treatments for all CF patients whatever their mutation. As the results of these and other new molecules appear, it is likely that a specific molecule or combination will be needed for each patient, depending on their existing mutations. In any case, the future is promising, and important steps have been taken toward obtaining a treatment that will effectively act on the cause of this disease.
Conflicts of InterestThe authors declare no conflicts of interest.
Please cite this article as: Quintana-Gallego E, Delgado-Pecellín I, Calero Acuña C. Tratamientos reparadores de la proteína CFTR en la fibrosis quística. Arch Bronconeumol. 2014;50:146–150.







