

Tuberculosis resistance diagnostics have vastly improved in recent years thanks to the development of standardised phenotypic and molecular testing methods. However, these methods are either slow or limited in the number of resistant genotypes they can detect. With the advent of next-generation sequencing (NGS) we can sidestep all those problems, as we can sequence whole tuberculosis genomes at increasingly smaller costs and requiring less and less DNA. In this review, we explain how accumulated knowledge in the field has allowed us to go from phenotypic testing to molecular methods to Whole Genome Sequencing (WGS) for resistance diagnostics. We compare current diagnostic methods with WGS as to their efficacy in detecting resistant cases, and show how forthcoming advances in NGS technologies will be crucial in widespread implementation of WGS as a diagnostic tool.
El diagnóstico de la tuberculosis resistente ha mejorado ampliamente en los últimos años gracias al desarrollo de pruebas estandarizadas de diagnóstico tanto fenotípicas como moleculares. Sin embargo, estas pruebas son o bien lentas o limitadas en el número de genotipos resistentes que son capaces de detectar. Con el auge de las nuevas tecnologías de secuenciación masiva podemos evitar esos problemas secuenciando el genoma completo cada vez a un coste más bajo y requiriendo cantidades menores de ADN. En esta revisión, explicamos cómo se ha podido progresar desde las pruebas fenotípicas a los métodos moleculares hasta la secuenciación del genoma completo para el diagnóstico de resistencias gracias a sucesivos descubrimientos en el campo. Comparamos la eficacia de la secuenciación del genoma completo para detectar casos resistentes con respecto a la de los métodos diagnósticos actuales, y mostramos cómo los avances futuros en esta tecnología serán cruciales para la implementación generalizada de esta herramienta diagnóstica.
Each year, over 1.5 million people die of tuberculosis all around the world.1 Once a neglected disease, it has received increasing attention in recent decades and its eradication by the year 2030 is one of the Sustainable Development Goals. However, this is unlikely to happen for several intertwined reasons, including widespread poverty in high-burden countries, low access to adequate healthcare, lack of an effective vaccine, and relevant to this review the spread of drug-resistant variants of the disease. Ever since the first documented drug-resistant tuberculosis case in the 1940s,2Mycobacterium tuberculosis has shown its ability to develop resistance to every single antitubercular drug (Table 1). However, drug-resistant tuberculosis did not become a global concern until the 1990s, when several outbreaks caused by resistant strains showed that such strains can indeed spread in the population.3 With around half a million new cases each year worldwide,1 drug-resistant tuberculosis has become an emerging threat that hampers our efforts to eradicate the disease. Meanwhile, there is a gap both in detection and in treatment of drug resistant tuberculosis cases with only 1 in 5 resistant cases being enrolled in the appropriate treatment.1
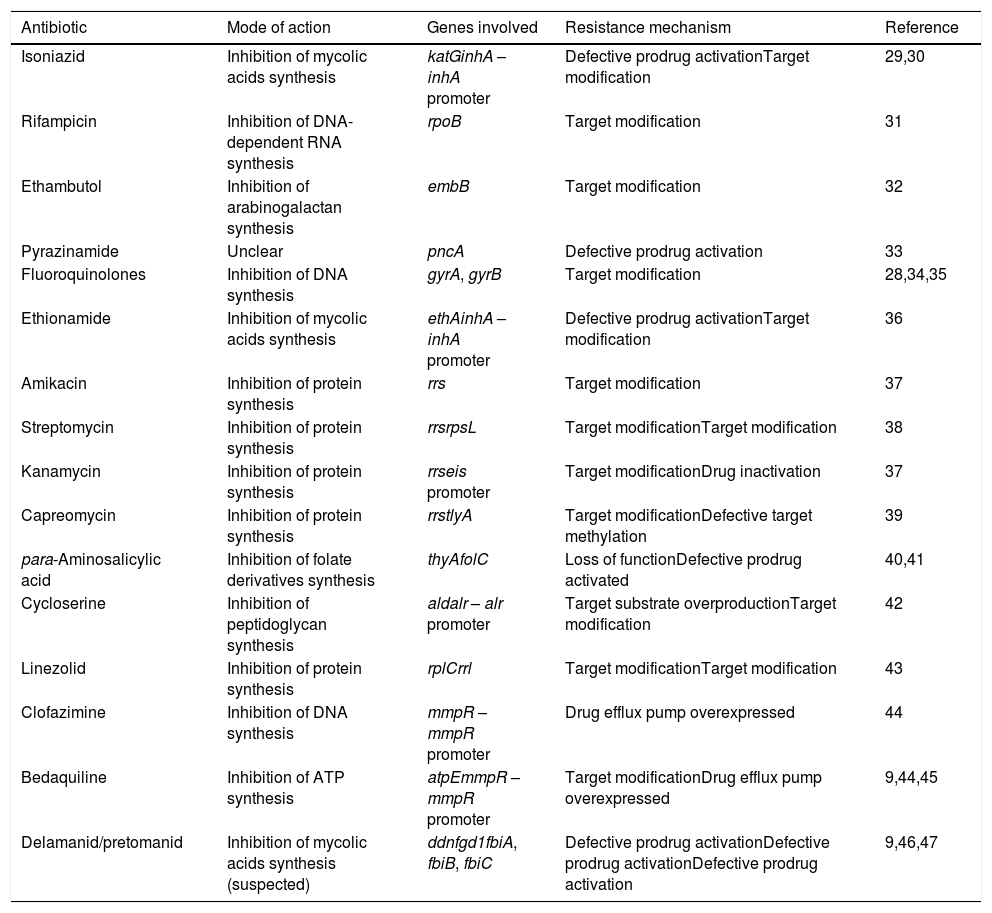
Modes of action and resistance mechanisms of antitubercular drugs in use.
| Antibiotic | Mode of action | Genes involved | Resistance mechanism | Reference |
|---|---|---|---|---|
| Isoniazid | Inhibition of mycolic acids synthesis | katGinhA – inhA promoter | Defective prodrug activationTarget modification | 29,30 |
| Rifampicin | Inhibition of DNA-dependent RNA synthesis | rpoB | Target modification | 31 |
| Ethambutol | Inhibition of arabinogalactan synthesis | embB | Target modification | 32 |
| Pyrazinamide | Unclear | pncA | Defective prodrug activation | 33 |
| Fluoroquinolones | Inhibition of DNA synthesis | gyrA, gyrB | Target modification | 28,34,35 |
| Ethionamide | Inhibition of mycolic acids synthesis | ethAinhA – inhA promoter | Defective prodrug activationTarget modification | 36 |
| Amikacin | Inhibition of protein synthesis | rrs | Target modification | 37 |
| Streptomycin | Inhibition of protein synthesis | rrsrpsL | Target modificationTarget modification | 38 |
| Kanamycin | Inhibition of protein synthesis | rrseis promoter | Target modificationDrug inactivation | 37 |
| Capreomycin | Inhibition of protein synthesis | rrstlyA | Target modificationDefective target methylation | 39 |
| para-Aminosalicylic acid | Inhibition of folate derivatives synthesis | thyAfolC | Loss of functionDefective prodrug activated | 40,41 |
| Cycloserine | Inhibition of peptidoglycan synthesis | aldalr – alr promoter | Target substrate overproductionTarget modification | 42 |
| Linezolid | Inhibition of protein synthesis | rplCrrl | Target modificationTarget modification | 43 |
| Clofazimine | Inhibition of DNA synthesis | mmpR – mmpR promoter | Drug efflux pump overexpressed | 44 |
| Bedaquiline | Inhibition of ATP synthesis | atpEmmpR – mmpR promoter | Target modificationDrug efflux pump overexpressed | 9,44,45 |
| Delamanid/pretomanid | Inhibition of mycolic acids synthesis (suspected) | ddnfgd1fbiA, fbiB, fbiC | Defective prodrug activationDefective prodrug activationDefective prodrug activation | 9,46,47 |
As things stand, we need faster, more reliable diagnostic methods of detecting resistance in tuberculosis. One possible solution would be to use our knowledge of the genetic basis of antibiotic resistance to predict the resistant phenotype using the nucleotide sequence. In recent years, we have started using a series of molecular testing methods that allow us to accurately detect diagnostic mutations for drug resistance.4 However, the catalog of mutations is limited and we need to be able to update it as we increase our knowledge in antibiotic resistance and having the actual nucleotide sequences would allow just that. What is more, with the advent of affordable high-throughput sequencing technologies, we could determine resistance in a fast and standardised way and on a massive scale.5 This way, we can finally reliably diagnose resistance while having the possibility of avoiding the time-consuming culturing step. In this review, we are going to (i) lay out our current understanding of the genetic bases of resistance; (ii) explain how we can use this knowledge to predict resistant strains; and (iii) make the case for the use of WGS as a routinary technique in resistance diagnostics in M. tuberculosis.
Antibiotic resistance in M. tuberculosis- a.
Drug resistance in M. tuberculosis: patterns and detection
It is estimated that around 6% of new tuberculosis cases are resistant.1 As drug resistance appears in response to treatment, it normally follows predictable patterns that allow us to optimise diagnostic resources. It first becomes resistant to the main first-line drugs, rifampicin and isoniazid. Then, after starting treatment with second-line drugs, the bacteria becomes resistant to them too and grows increasingly untreatable. This process may occur within a single patient (see Ref. 6 for a particularly well-documented example) or could take place in multiple successive infections in which resistant bacteria are transmitted from one patient to another, acquiring further resistance mutations along the way. Although transmission of resistant variants was initially dismissed as unlikely, recent data suggest that such transmission is ongoing and probably increasing.1 Transmission of drug-resistant variants can hamper eradication efforts, particularly in high-burden countries. In a worst-case scenario, we could end up in a situation in which susceptible variants are progressively being substituted by resistant ones, drastically reducing our ability to treat tuberculosis.7 Thus, being able to detect, track and stop the spread of resistance is of capital importance for an effective control of the disease.
Tuberculosis diagnostics, including resistance testing, has traditionally required specific expertise and infrastructure. The standard method is to directly measure the phenotype using a drug susceptibility test (DST). This can be done using both liquid and solid cultures and depending on how it is performed it can also show the specific level of resistance of the strain that is being tested. In recent times standardised kits have been developed to reduce workload and increase speed and reproducibility. Becton Dickinson's BACTEC MGIT system8 is one of the most widely used for its reliability and ease of use but is only available for some antibiotics and requires expensive equipment and reagents. Thermo Scientific's Sensititre MYCOTB plate allows to perform accurate minimal inhibitory concentration (MIC) determinations to the most important first- and second-line antibiotics in a standardised way, but is also expensive and requires technical expertise. Although DST is reliable and is the only way that you can actually determine whether a strain is sensitive or resistant, it is also slow and difficult to perform, and provides no information beyond the phenotype. This makes DST unsuitable as the only method for drug resistance control in high burden countries where transmission is ongoing and resistant outbreaks need to be controlled fast, and in countries where resources are limited.
As sequence information for resistant strains started to accumulate, a clear pattern linking drug resistance mainly to a defined set on single-nucleotide mutations emerged, meaning that resistance could be confidently predicted based on sequence alone. Following this line of reasoning, a myriad of new molecular testing methods emerged, all of them using a PCR approach to determine resistance to a series of first- and second-line antibiotics. Although resistance prediction directly from sequence information is possible and has been useful in detecting resistant variants for antibiotics with no commercial test,9 it is still not accepted as standard practice.
- b.
Tuberculosis treatment, targets and resistance
Tuberculosis treatment is very standardised and is always tailored to the bacterial phenotype. One of the main features of the bacteria is its mycolic acid wall, which makes it intrinsically resistant to some compounds such as beta-lactam antibiotics.10 All sensitive cases are routinely treated with the two main first-line antibiotics, rifampicin and isoniazid, in combination with ethambutol and pyrazinamide. When the bacteria become resistant to both rifampicin and isoniazid, it is assumed to be a multi-drug resistant case (MDR-TB) and the treatment must then incorporate second-line antibiotics such as fluoroquinolones and injectable aminoglycosides among others. When MDR-TB additionally acquires resistance to both at least one fluoroquinolone and one injectable drug, it is considered an extensively drug resistant case (XDR-TB) and the treatment must then incorporate add-on second-line antibiotics. When resistance appears as a consequence of normal antibiotic treatment it usually follows this predictable pattern. In any case, drug-resistant tuberculosis can still be effectively treated, but it is necessary to know the exact sensitivity profile of the infecting strain to be able to provide a tailored combination of antibiotics. Last August and after reviewing the latest evidence available, the World Health Organisation (WHO) decided to eliminate injectable antibiotics from recommended second-line treatments and promote the use of bedaquiline to treat all MDR-TB cases.11 These changes are a first step to eventually obtain a short-course all-oral treatment which would be positive for drug resistance control, but has the drawback of commercial molecular tests for bedaquiline resistance not being available yet. Furthermore, the genetic basis of bedaquiline resistance is not fully understood yet, so the full implications of this change are unknown.
- c.
The importance of single mutations
M. tuberculosis is a highly clonal bacterial species and experiences no lateral gene transfer. As a consequence, drug resistance is acquired mainly by single mutations that either modify the target or interfere with prodrug activation.12 Furthermore, in many cases the genes involved are either essential or at least very important for bacterial survival and propagation. This means that even if a mutation confers resistance it cannot be too detrimental as well or it will be easily outcompeted by other mutations.13 For this reason, most of the resistance mutations that we observe are recurring even when other mutations are possible, as it is the case with isoniazid or rifampicin, where particular resistance mutations are present in most of the isolates (katG S315T and rpoB S531L/S441L respectively).14 This recurrence implies that phenotypic resistance will most probably be tied to a small subset of mutations and we will be able to predict a resistant phenotype when we see a particular genotype. We must be cautious, however, as the predictability of resistance mutations is highly antibiotic-dependent: for instance, a high amount of pyrazinamide resistance mutations in the pncA gene found in isolates have never been described before, but phenotypic analyses confirm that they are conferring resistance.15
WGS for resistance diagnostics- a.
Use of mutations as resistance markers
In recent years, we have seen a rapid surge of molecular resistance testing methods. Phenotypic testing is slow, labour-intensive and requires specific infrastructure, which delays diagnostic at the expense of patients. Molecular methods, on the contrary, offer fast diagnosis, standardised and potentially high-throughput testing and fewer biosafety requirements but are more expensive and depend strongly on our knowledge of the genetic determinants of resistance. For these reasons, although WHO endorses some of those tests they still have not replaced DST.4
Commercially available molecular tests are all nucleic acid amplification tests, in which PCR is used to detect specific nucleotide sequences, but they use different methods of detection and are not used for the same antibiotics. The GeneXpert and GeneXpert ultra systems (Cepheid) are used to detect rifampicin-resistant tuberculosis directly from sputum samples using ultra-sensitive PCR. These systems use probes to detect resistant variants directly while amplification is ongoing, thus reducing diagnosis time. Line probe assays use reverse hybridisation methods to detect resistant variants to first- and second-line antibiotics. These methods are fast, reliable and accurately predict resistance in 88–95% of the cases for rifampicin, 91% for isoniazid and 86–100% for second-line antibiotics.16 However, they are necessarily constrained to specific sets of mutations and thus lack flexibility. For instance, there is no line probe test available for pyrazinamide because the set of resistance mutations for this antibiotic is both big and incomplete as apparently any mutation that modifies pncA gene can render the bacteria resistant. Another drawback of molecular testing methods is that they do not provide any additional information on other mutations in the genome, which would be valuable to further understand the genetic basis of antibiotic resistance or to understand how it is affected by the genetic background.
- b.
Next-generation sequencing as a diagnostic tool
Next-generation sequencing (NGS) refers to an array of technologies used to massively sequence DNA fragments. Today, widely used NGS platforms are based on short reads with low error rates that allow to obtain high-confidence variant calls.17 This reliability has made NGS a powerful new tool for the diagnosis of multiple genetic diseases18 and infections.19 In the particular case of M. tuberculosis, its small bacterial genome is the perfect candidate for this kind of technology as a diagnostic application.20 Whole genome sequencing (WGS) refers to obtaining the complete genomic sequence of an organism in a single sequencing run and it can be done to study virtually every strain genome isolated in the clinical microbiology units. This has led to several health agencies in the world to study the role of WGS as a diagnostic tool for drug resistance prediction. The first major publication using WGS was by Walker et al.,5 demonstrating that, based on a known list of drug resistance associated mutations, high sensitivity and specificity values could be achieved for rifampicin and isoniazid. But WGS does not constrain us to any particular set of resistance mutations and also allows us to come back and re-analyse the sequence afterwards. Furthermore, we can use the whole genome sequence to trace the epidemiological network of the strain.21 Thus, if we were able to use genomic sequence information in a systematic way we would be able to both detect resistances more accurately than with current molecular assays and obtain valuable data to understand the arisal and spread of drug resistance.
A simplified pipeline for WGS of tuberculosis would be: sample collection, culture, DNA extraction, library preparation, sequencing and bioinformatic analysis for variant detection which allows resistance prediction (Fig. 1). Right now our best predictions come from rifampicin and isoniazid to the point that it has been recently shown that susceptibility to those drugs can be accurately predicted.22 This result has led to major health institutes (NHS England, Netherlands, New York State) to fade down phenotypic testing in favour of WGS and only perform detailed phenotypic analyses when the patient is not responding or when resistance to first line drugs are detected by WGS. Thus this recent publication represents a copernican shift on how tuberculosis resistances are diagnosed. It is however important to note that culture is still vital if we want to link new variants to drug resistance, as in high-burden countries with MDR-TB sequencing capacities are still limited and expensive. However there is hope that sequencing the genome from the sputum sample will be possible soon and we will do it with cheaper and portable technologies such as MinION (Oxford Nanopore Technologies).23 Thus while our catalog of drug resistance associated variants is expanding everyday so does it our ability to establish genome sequencing technologies at the point of care in places where tuberculosis represents a major health and economic burden.
- c.
Taking it further: comprehensive resistance mutations databases

A simplified workflow for WGS in tuberculosis. After sample collection from the patient, usually a sputum, bacteria are cultured in plates during 3–4 weeks until their number is sufficient enough to obtain a good amount of DNA through extraction. This DNA then undergoes a library preparation protocol until it is ready for the Illumina sequencing platform of choice. The final bioinformatic analysis usually consists on mapping the obtained reads to a reference sequence and calling variants. If these variants are present in resistance databases, predictions are generated for the isolate.
With the advent of WGS as a diagnostic tool, there is a need to accurately interpret the large amount of data coming from this technique. In short, common analysis pipelines consist on mapping the sequenced samples to a reference genome and calling variants that are annotated using a database. Due to the importance of separating mutations that confer resistance to antibiotics from those that do not, there has been considerable effort in generating such databases in recent times. We must build reliable, flexible and evidence-based databases to overcome the challenge of resistance annotation. This stems from the need to correctly diagnose patients, and we should be moving towards a standardised annotation protocol using centralised and curated databases.
- i.
How a resistance mutations database is built
The first approaches at building resistance mutations databases were a community effort, where common mutations found independently at several labs were linked to a resistance phenotype. In the last years, several studies have tried to improve classic databases by analysing big datasets and trying to associate recurring SNPs in isolates to phenotypic resistance in several ways such as searching known genes for new variants.5,24 More recently, other studies have devised a GWAS approach scanning global collections of strains for known and new mutations and then performing standardised DST to try to establish genotype-phenotype strong associations.15 Finally, international consortiums of independent experts have created platforms to gather and share sequence and DST data in order to build a reliable database for common and rare mutations. These will be of utmost importance, as several studies in non-developed countries25 begin to claim that resistance detection using current databases are systematically failing due to rare mutations arising in these low antibiotic pressure settings. We have reached the phase where we need to work with huge and representative datasets to get a reliable mutation database that can predict resistance regardless the origin of an isolate.
- ii.
Different databases, similarities and differences
One of the strong points for generating mutation databases is to ease the detection of resistance-associated variants in a clinical setting without requiring bioinformatic knowledge in NGS data analysis. From the many databases that have been built, some of them offer the option to analyse your sequence data in a completely independent and automated manner. This is the case of PhyResSE (http://www.phyresse.org), TBProfiler (http://tbdr.lshtm.ac.uk) or CASTB (http://castb.ri.ncgm.go.jp/CASTB/), web-based tools which do not require any bioinformatic knowledge by the user and will generate reports detailing the predicted lineage and resistance profile of the uploaded data. These services feature different databases for their predictions, for instance PhyResSE and CASTB are built entirely from literature mutations while TBProfiler is the result of a global literature dataset analysis. There are also downloadable simple programs to perform predictions, such as Mykrobe Predictor TB (http://www.mykrobe.com/products/predictor) that do not require any expertise to be used either.
However, the most promising databases will surely be originated from consortiums like ReSeqTB (https://platform.reseqtb.org) or CRyPTIC (http://www.crypticproject.org) because the current ones are quite biased to common mutations that mostly appear in first-world countries where the treatment regimes are followed strictly. A global strain collection coupled with standardised DST approach seems to be the most robust method to identify new region-specific variants. Consortiums such as those are vital, because while others are just simply collecting information, they are actively generating knowledge. A database is as good as the data it was built with and how representative it is, so these initiatives will play a big role in the coming years.
- d.
Limitations: automation, costs, sensitivity and specificity
Whole genome sequencing for resistance detection is sure to become a standard practice in the near future over other traditional techniques in diagnosis, but we must be aware of its limitations as of today. First, automation of library preparations are limited to big research facilities having robotic arms.26 Otherwise, specialised staff is required to achieve good results in sequencing, as library quality determines how good the sequencing results you get are. The bioinformatic analysis of the sequences, on the other hand, is a process that is being automatised nowadays, although there is still work to do around the standardisation of these analysis pipelines over different research labs before they can become a solid clinical practice. Second, although the cost of sequencing has dramatically lowered in the last years, WGS for resistance detection is still overall more expensive than other techniques. It depends on several factors like the sequencing platform, the kit used or the read depth desired, but the estimated cost for a single isolate can be situated around 150–200€.27 Finally, the big challenge is reaching high values of sensitivity and specificity in our predictions using mutation databases. Although those are relatively high for isoniazid (90%, 97%), rifampicin (93%, 98%) and ethambutol (90%, 92%), pyrazinamide is not so well-characterised (65%, 97%). Also, resistances to second-line drugs are more difficult to predict, for instance amikacin (85%, 93%), moxifloxacin (85%, 90%), d-cycloserine (61%, 90%) or PAS (65%, 94%), according to recent studies.15 While there are phenotypic testing problems with drugs such as ethambutol and pyrazinamide, others like fluoroquinolones (FQ) are easier to test. However, it is now clear that not all mutations traditionally associated to FQ resistance lead to antibiotic resistance in the three FQ used in the clinical setting.28 This is a key result as some mutations are specific to moxifloxacin leaving room to use another FQ without changing the regimen of the patient. As new and repurposed drugs are being increasingly used in patients with no other therapeutic option our capacity to predict the DST of the strain from WGS decreases. This limitation comes from the scarce data, even from phenotypic testing, on drug resistant strains to those drugs and will likely ease in the coming years with increasing use of the drugs and genome sequencing of resistant cases. Thus a key player in WGS diagnostics is our available catalogue of curated mutations for each drug.
ConclusionsWGS is well on its way to become a major diagnostics tool in tuberculosis. Its main advantages are that it allows to probe the whole genome for resistance mutations and can be used retrospectively. Nowadays, it has already shown to be effective in detecting 98% of pansusceptible strains in a recent study,22 so it may in the future eliminate the necessity for routinely performing phenotypic testing on them. On the other hand, it still has a long way to go on standardisation and reducing costs. Additionally, it works best on first-line antibiotics but needs improvement on diagnostics for second-line drugs. Hopefully, as ongoing WGS projects obtain more data we will be able to accurately predict resistance for most antibiotics. If used correctly, WGS resistance diagnostics would decisively help us to provide truly personalised care to any patient.
FundingThe authors were supported by the project SAF2016-77346-R, awarded to Iñaki Comas and the grant BES-2017-079656, awarded to Miguel Moreno-Molina by the Spanish Ministry of Economy and Competitiveness. The grant provider played no part in study design, data collection and analysis, or the preparation of the manuscript.
Conflict of interestsThe authors declare no conflict of interests.







