



Introducción
La inflamación es una reacción inespecífica del tejido conjuntivo vascular que actúa como respuesta protectora del organismo frente a diversas noxas1,2. Consta de una reacción inicial o aguda (eslabones 1 y 2) y de una reacción tardía (eslabón 3):
Eslabón 1. Frente a una noxa, los elementos correspondientes a la inmunidad natural, que existen perennemente en sangre (neutrófilos, células citolíticas, complemento, entre otros), se activan y liberan sustancias que dan lugar a los procesos básicos de este eslabón: edema, vasodilatación y llegada de neutrófilos.
Eslabón 2. Una vez activados los diferentes elementos que corresponden a la respuesta inicial, se produce el rodamiento de diversas estirpes celulares en el endotelio y se generan diversas moléculas que favorecen su adhesión y migración a los tejidos; son las llamadas moléculas de adhesión, que permiten y favorecen el paso de leucocitos a la membrana basal y tejidos. Asimismo, el endotelio genera mediadores vasoactivos (óxido nítrico, prostaciclina, endotelina-1 y tromboxano), que regulan el tono vascular.
Eslabón 3. En los tejidos los neutrófilos y macrófagos emigrados tienen múltiples acciones, ya sean bactericidas o de liberación de sustancias que, a su vez, pueden inducir lesiones vasculares a través de la dificultad para generar mediadores vasoactivos como el óxido nítrico y la generación de múltiples mediadores que activan diversas vías de la inflamación, se retroalimentan e implican a sistemas celulares muy diversos (linfocitos, entre otros).
En el síndrome de apneas e hipopneas durante el sueño (SAHS), uno de los elementos patogénicos más importante en la cascada inflamatoria es la producción de radicales libres.
Vía aérea
En la última década han aparecido datos en pacientes con SAHS que revelan la presencia de marcadores de inflamación en la vía aérea superior (VAS) e indican que la inflamación podría tener un papel en la fisiopatología del SAHS3-9. En estos pacientes es frecuente el hallazgo macroscópico de inflamación local, aumento del tamaño y enrojecimiento de la úvula y del paladar blando. Se postula que estos cambios inflamatorios locales se deben a la vibración generada por el ronquido. Pero no sólo se han encontrado cambios inflamatorios en las estructuras susceptibles a la vibración del ronquido, sino que los pacientes con SAHS, en ausencia de otros procesos que lo justifiquen, tienen inflamación nasal4, indicios de inflamación en la vía aérea3,5, aumento de neutrófilos en el esputo inducido e incluso inflamación en el parénquima pulmonar5, lo que también se ha observado en un modelo de SAHS en rata7. Se ha señalado que, además del ronquido, las presiones muy negativas generadas a lo largo de todo el sistema respiratorio durante las obstrucciones podrían contribuir a la aparición de inflamación3. Los estudios histológicos de la VAS en el SAHS han descrito la presencia de un importante edema subepitelial y un aumento del grosor de la lámina propia3,8. Conjuntamente con el edema, se han señalado otras alteraciones histopatológicas en la VAS de pacientes con SAHS, donde la inflamación parece tener un papel. Sekosan et al9 describieron la aparición de un extenso infiltrado inflamatorio leucocitario con predominio de células plasmáticas en la lámina propia. Por su parte, Paulsen et al3 detectaron un infiltrado inflamatorio en la lámina propia, en este caso con predominio de células T (CD3+), junto con una reducción de la densidad y altura de las estructuras papilares del tejido conectivo que se introducen hacia el epitelio y cuya función es el anclaje estructural y soporte nutricional. En este estudio, los pacientes con SAHS presentaron diferencias en el patrón de expresión de algunas citoqueratinas y acantosis en el epitelio. También se encontraron cambios estructurales, como la acantosis en el epitelio, hipertrofia en las glándulas mucosas, atrofia focal de fibras musculares y cambios degenerativos en nervios periféricos. En este sentido, también se han descrito anomalías en las terminaciones nerviosas musculares, con evidencia de denervación, acompañada de un infiltrado inflamatorio con predominio de células T (CD4+ y CD25+) y numerosas terminaciones nerviosas anómalas de aspecto varicoso en la lámina propia.
Todo ello hace pensar en la presencia de una neuropatía, probablemente mediada por inflamación, que sería responsable de los déficit sensoriales que se observan en la VAS del SAHS y de la disfunción muscular de la VAS, que contribuirán al proceso de obstrucción. Algunas de estas alteraciones también se han identificado en pacientes sin apneas que roncan, aunque en un grado menor, pero no en individuos controles que no roncan. Cabría pensar entonces que en las fases iniciales la vibración del ronquido provocaría inflamación local en la VAS, que iría progresando paulatinamente. Aparte del compromiso estructural que presentan estos pacientes, la inflamación local desempeñaría un papel en la disfunción neuromuscular de la VAS alterando los reflejos y la función de los músculos que controlan el calibre de la vía aérea. Una vez que los eventos respiratorios obstructivos están presentes, los cambios en las presiones de la vía aérea y la hipoxemia contribuirían a amplificar la inflamación a otras zonas del aparato respiratorio, incluso a nivel sistémico. Se entraría en un círculo vicioso, en el que a más inflamación, mayor disfunción, más obstrucción y más presiones negativas, que finalmente generarían más inflamación.
SAHS, disfunción vascular e inflamación
La relación entre el SAHS y la enfermedad cardiovascular puede producirse por diversos factores: cambios mecánicos, neurovegetativos e inflamatorios.
Cambios mecánicos
Las presiones intratorácicas muy negativas, que pueden llegar a 100 cmH2O, provocan un aumento del gradiente transmiocárdico y, en consecuencia, aumenta de un modo considerable la poscarga. Estas presiones negativas dan lugar a un aumento del retorno venoso, que desplaza el septo hacia la izquierda, con el consiguiente deterioro de la función del ventrículo izquierdo. Así pues, la combinación del aumento de la poscarga por las presiones negativas y también por la hipertensión que se genera, el desplazamiento del septo y, finalmente, la hipoxemia que se crea dan lugar a una disfunción cardíaca que revierte totalmente con presión positiva continua de la vía aérea10,11.
Anomalías neurovegetativas
En personas sanas, durante la noche la actividad simpática, la presión arterial y la frecuencia cardíaca diminuyen; en la fase REM, aumentan. Los quimio y barorreceptores controlan los cambios gasométricos y de la presión arterial a través, entre otros, de la activación o reducción de la actividad del sistema simpático y parasimpático. Los pacientes con SAHS, por la hipoxemia y el resto de eventos que ocurren durante las apneas, muestran un incremento notorio de la actividad simpática durante la noche y durante el día que incrementa la presión arterial. En comparación con individuos sanos, presentan taquicardia, reducción de la variabilidad cardíaca y aumento de la variabilidad de la presión arterial. Todos estos cambios representan un factor de riesgo acusado de daño vascular12.
Cambios inflamatorios e incremento de radicales libres
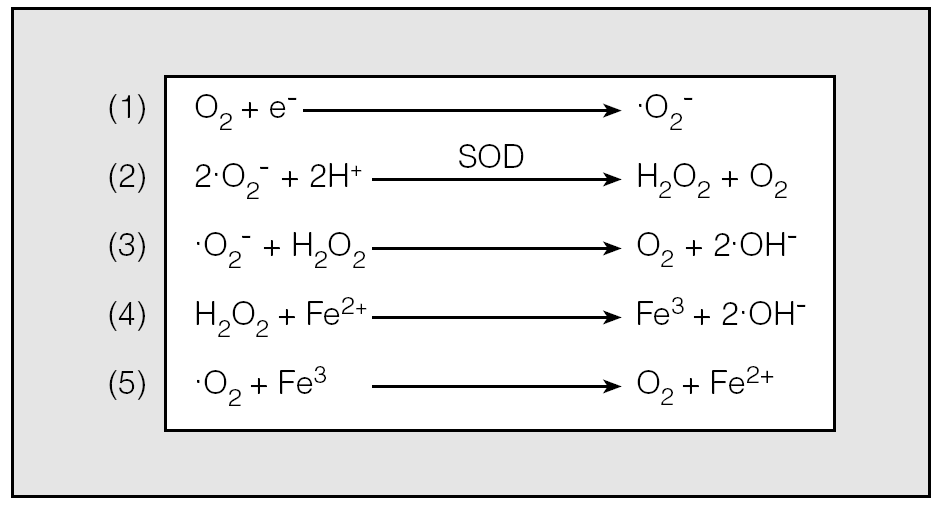
En el SAHS uno de los elementos que se han considerado más importantes para el inicio de una reacción inflamatoria sistémica es la excesiva producción de radicales libres13-15. Éstos son moléculas con uno o más electrones desapareados, que tienen una función fisiológica en el sentido de intervenir en el metabolismo celular para la obtención de energía. Intervienen en la formación de adenosintrifosfato y H2O. También auxilian a los neutrófilos para la lisis de noxas (tabla I). En la figura 1 se resumen las diversas reacciones para la producción de radicales libres. Dado que en sí mismos son tóxicos, cuando se produce un aumento de radicales libres, hay unos mecanismos de defensa (antioxidantes) (fig. 2) que los neutralizan. Cuando, por un exceso de radicales libres, éstos no son neutralizados, se liberan y se desencadena un proceso de estrés oxidativo que inducirá daño celular (fig. 3).

Fig. 1. Reacciones de generación de los 3 principales radicales libres de oxígeno: el ion superóxido (O2), el peróxido de hidrógeno (H2O2) y el radical hidroxilo (OH·). El oxígeno molecular puede aceptar un electrón y generar así el radical superóxido (1). La interacción entre 2 radicales superóxido, catalizada por la superóxido dismutasa (SOD), conduce a la formación de peróxido de hidrógeno(2). El radical hidroxilo se forma por la interacción, mediada por hierro, entre el peróxido de hidrógeno y el radical superóxido(3), o bien por la interacción directa del peróxido de hidrógeno con iones Fe+2 (4). En presencia de superóxido, el Fe3+ unido a la ferritina se libera como Fe2+, con lo que aumenta la disponibilidad de Fe2+ para estas reacciones(5). Dado que la SOD y los iones hierro están fácilmente disponibles y en abundancia en los sistemas biológicos, las reacciones(2, 4y5)se producen de forma simultánea. En consecuencia, la formación del radical superóxido comporta la rápida producción de peróxido de hidrógeno y del radical hidroxilo.


Fig. 2. Los sistemas de defensa del organismo frente al estrés oxidativo (antioxidantes) se dividen en enzimáticos y no enzimáticos. En los mecanismos enzimáticos, las enzimas implicadas se agrupan en 3 grupos: superóxido dismutasas, catalasas y perioxidasas. Hay también una serie de sistemas de defensa no enzimáticos que se componen de sustancias con capacidad de reaccionar directamente con los radicales libres, o bien con productos de éstos sin necesidad de ninguna enzima. Entre estos antioxidantes se encuentran las vitaminas C y E, los betacarotenos, el ácido úrico y los flavonoides.

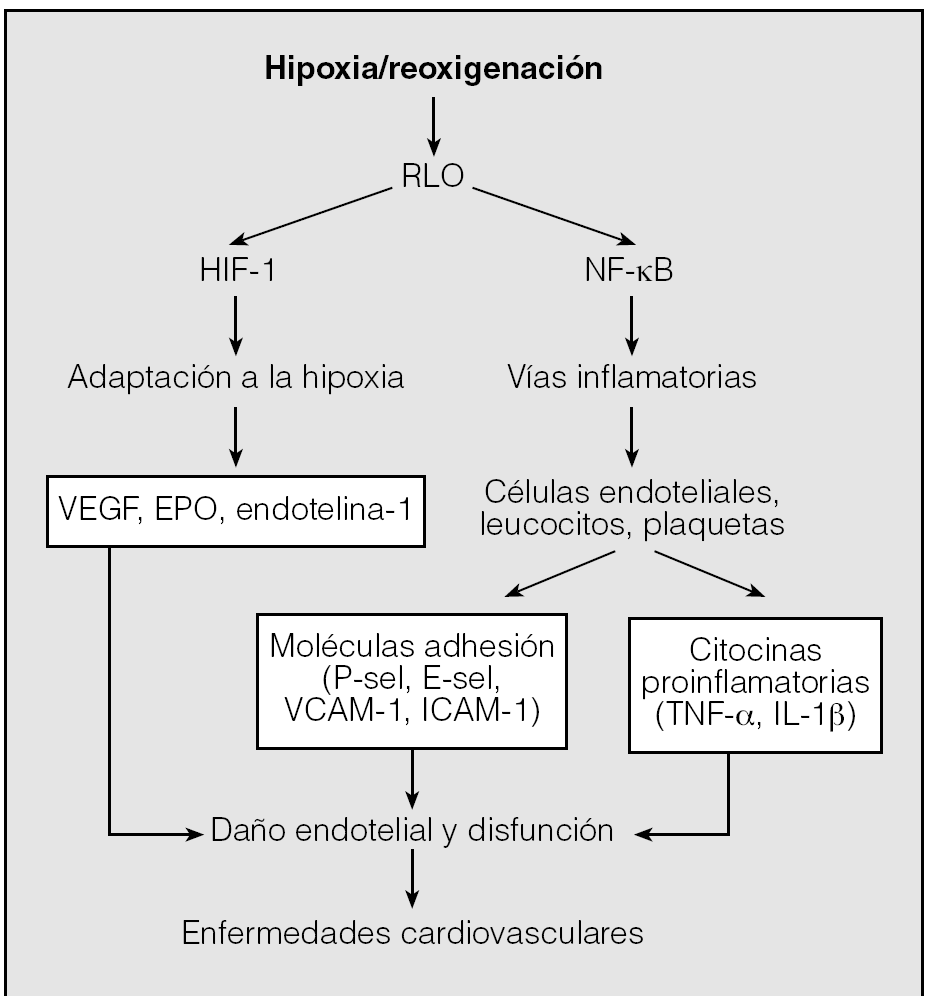
Fig. 3. Esquema de los mecanismos que intervienen en la apnea obstructiva del sueño y desencadenan enfermedades cardiovasculares. En respuesta a la hipoxia/reoxigenación, la liberación de radicales libres de oxígeno (RLO) da lugar a la activación de los factores de transcripción. El factor de transcripción kβ (NF-κB) da lugar a la transcripción de diferentes citocinas inflamatorias, y el factor inducible por hipoxia (HIF-1) da lugar a la transcripción de factores de crecimiento y otras citocinas en relación con la hipoxemia. Conjuntamente los productos génicos obtenidos de estos 2 factores de transcripción generarán un daño endotelial y su posterior disfunción, lo que producirá enfermedades cardiovasculares. EPO: eritropoyetina; E-sel: E-selectina; ICAM-1: moléculas de adhesión intercelular-1; IL: interleucina; P-sel: P-selectina; TNF: factor de necrosis tumoral; VCAM-1: moléculas de adhesión vascular-1; VEGF: factor de crecimiento del endotelio vascular.
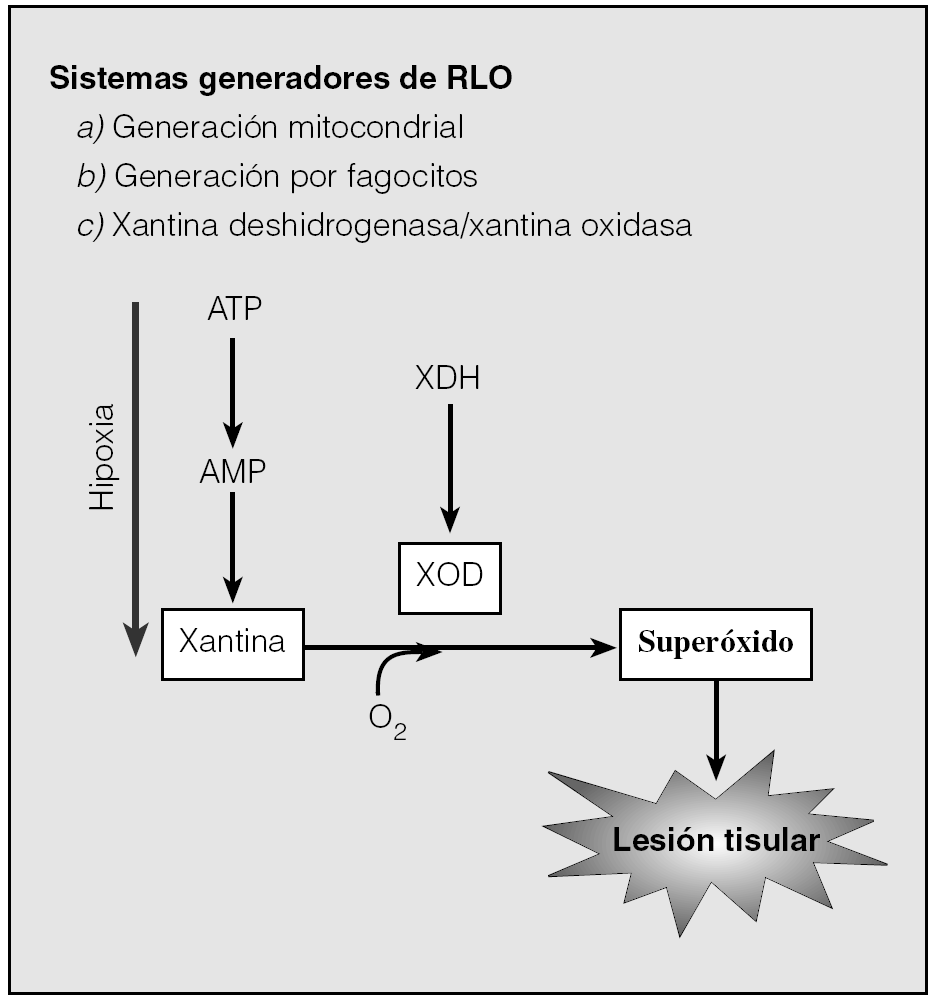
Los radicales libres circulan por el organismo intentando robar un electrón de las moléculas estables, con el fin de alcanzar su estabilidad electroquímica. Una vez que el radical libre ha conseguido robar el electrón que necesita para aparear su electrón libre, la molécula estable que se lo cede se convierte a su vez en un radical libre, al quedar con un electrón desapareado; de este modo se inicia una verdadera reacción en cadena que destruye o deteriora células. En la figura 4 se muestran los tres sistemas generadores de radicales libres. Durante la apnea, como consecuencia de la hipoxia/normoxia, que puede considerase una situación muy particular, se afectan las vías metabólicas celulares y se producen intensas disfunciones celulares que provocan un elevado número de radicales libres, con lo que se desencadena estrés oxidativo.

Fig. 4. Generación de radicales libres de oxígeno (RLO). Durante el proceso de hipoxia, la falta de aporte de oxígeno al tejido hace que se detenga el transporte de electrones en la mitocondria y, como consecuencia, disminuyen los valores energéticos en las células. Se producen cambios en los sistemas xantina deshidrogenasa (XDH)/xantina oxidasa (XOD), que después, frente a la hiperoxia, sufren cambios que llevan a la formación de radicales libres. AMP: adenosinmonofosfato; ATP: adenosintrifosfato.
Efectos nocivos
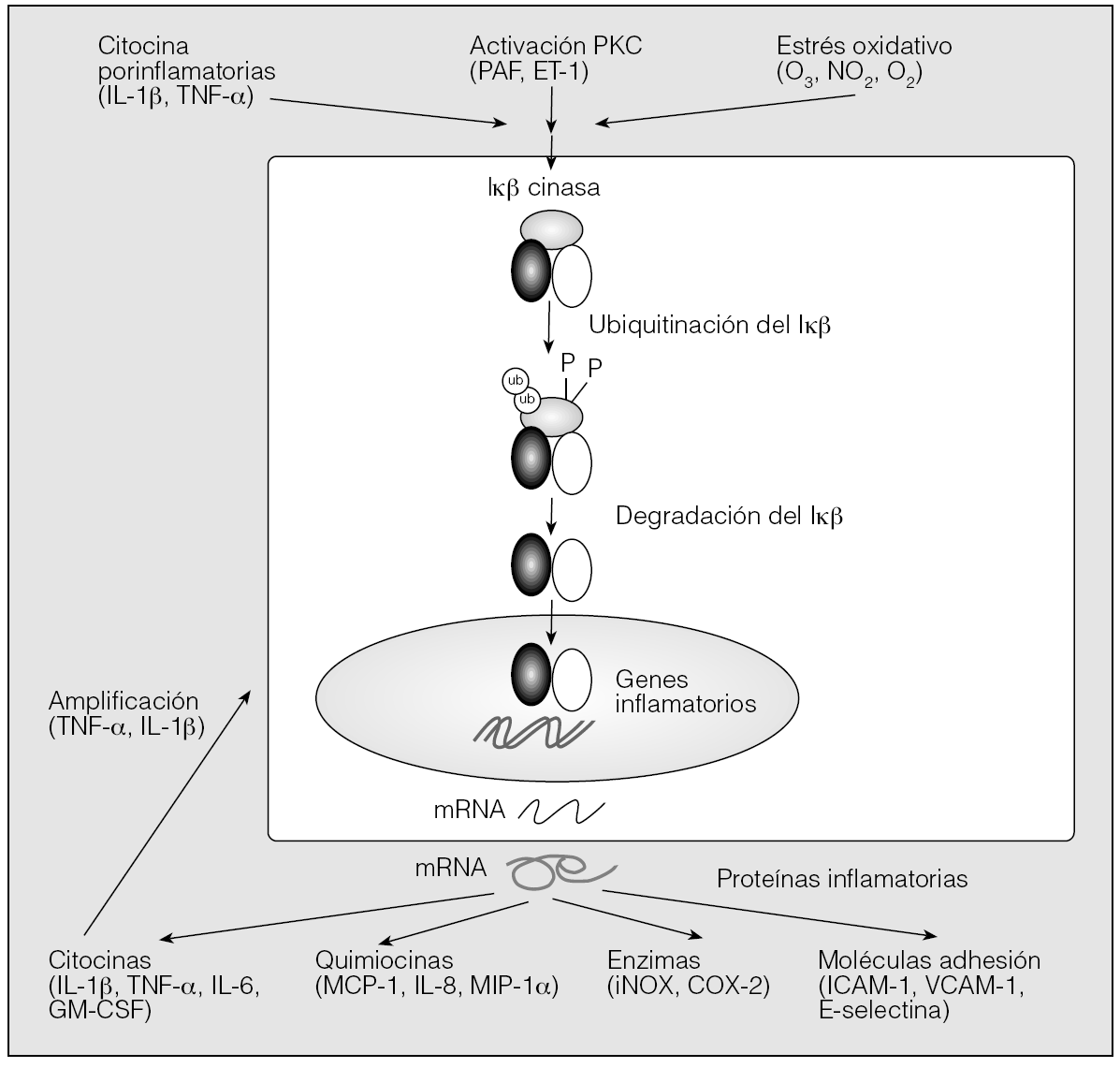
En la figura 3 se muestra un esquema general de las vías que se activarán como consecuencia del estrés oxidativo. Se desencadenarán cascadas de transducción de señales, que estimularán la secreción del factor de necrosis tumoral alfa (TNF-α), del factor activador de las plaquetas y de diferentes interleucinas (IL). Pueden actuar de una manera directa sobre el tejido afectado y producir la oxidación de lipoproteínas de baja densidad, la pérdida de viabilidad del óxido nítrico y la respuesta inflamatoria vascular, que desempeñan un papel crítico en la patogenia de la disfunción endotelial. Tal como se muestra en las figuras 3, 5 y 6, se activan factores de transcripción sensibles a redox que intervienen en el SASH, como el factor nuclear κB (NF-κB) y el factor inducible por hipoxia (HIF-1)16-18, que podrán desencadenar lesión endotelial13-15.

Fig. 5. Esquema representativo de la activación del factor nuclear κB (NF-κB) y de los genes que regula. Bajo un estímulo extracelular, como pueden ser citocinas como el factor de necrosis tumoral alfa (TNF-α) o la interleucina (IL) 1 y sobre todo el estrés oxidativo, se activa el complejo IkB cinasa (IKK). Este complejo media la fosforilación de la IκB, lo que da lugar a su ubiquitinación y degradación, dejando el dímero de NF-κB libre para translocar al núcleo, donde activará genes diana específicos a través de uniones selectivas a la secuencia consenso del NF-κB. Se producen diversas proteínas implicadas en la inflamación. ARNm: ARN mensajero; COX-2: ciclooxigenasa-2; ET-1: endotelina-1; GM-CSF: factor estimulante de colonias granulocíticas y microcíticas; ICAM: moléculas de adhesión intercelular; iNOS: óxido nítrico sintasa inducible; MCP-1; proteína quimiotáctica de los monocitos-1; MIP-1α: proteína inflamatoria de macrófagos-1α; PAF: factor activador de plaquetas; PKC: proteincinasa C; VCAM: moléculas de adhesión vascular.

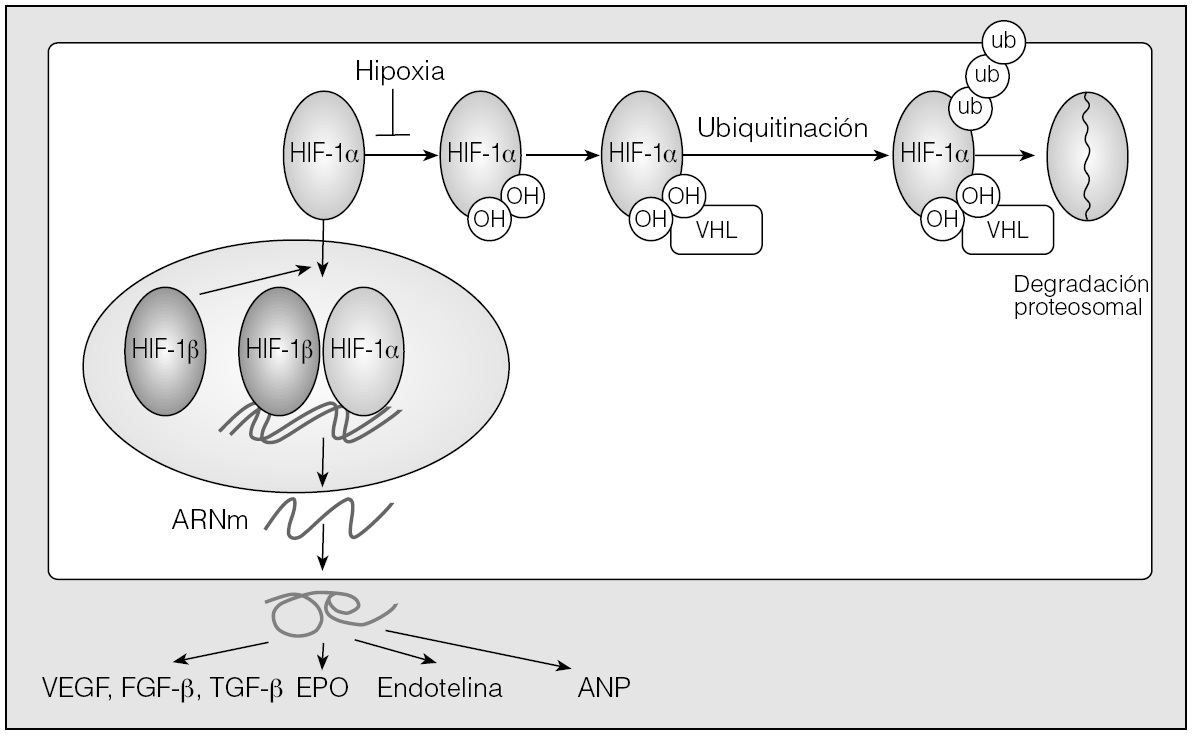
Fig. 6. Esquema representativo de la activación del factor inducible por hipoxia (HIF-1) y de los genes que regula. El HIF-1 está constituido por 2 subunidades, la alfa y la beta. En condiciones de normoxia, el HIF-1α es hidroxilado y posteriormente es reconocido por la proteína de Von Hippel-Lindau (VHL), la cual a su vez es señal para formar un complejo de ubiquitinación, que es indicativa de degradación por proteosomas. Sin embargo, en condiciones de hipoxia las hidroxilasas dejan de modificar al HIF-1α y éste se acopla al HIF-1β para ser translocado al núcleo, donde funciona como factor de transcripción. ANP: péptido natriurético auricular; ARNm: ARN mensajero; EPO: eritropoyetina; FGF: factor de crecimiento fibroblástico; TGF: factor transformador del crecimiento; VEGF; factor de crecimiento del endotelio vascular.
Sistemas de defensa ante el estrés oxidativo (antioxidantes)
Se resumen en la figura 2. Se dividen en enzimáticos y no enzimáticos. En los mecanismos enzimáticos, las enzimas que participan en la defensa contra la toxicidad del oxígeno pueden dividirse en 3 grupos: superóxido dismutasa, catalasa y peroxidasa. Además hay una serie de sistemas de defensa no enzimáticos. Las células disponen de un número elevado de sustancias con la capacidad de reaccionar directamente con los radicales libres o bien con productos de éstos sin necesidad de ninguna enzima. Entre estos antioxidantes se encuentran las vitaminas C y E, los betacarotenos, el ácido úrico y los flavonoides19.
Apnea obstructiva del sueño y vías de señalización molecular de los factores de transcripción
Factor nuclear-κB (NF-κB)
Se muestra en las figuras 3 y 5 con más detalle. El NF-κB es un factor de transcripción que regula la expresión de diferentes genes, incluidas citocinas, quimiocinas, factores de crecimiento, moléculas de adhesión y algunas proteínas20. Desempeña un papel muy importante en las respuestas celulares a diferentes tipos de agresiones, ya que regula la expresión inducible de genes que intervienen en la respuesta inmunitaria y el proceso inflamatorio21. En las células no estimuladas, el NF-κB se encuentra en el citoplasma en forma inactiva, asociado a una proteína que lo inhibe, llamada proteína inhibidora κB (κB), que impide la translocación del NF-κB al núcleo cuando no hay señal de activación.
Cuando la célula recibe una señal de activación, la IκB es fosforilada y degradada rápidamente por enzimas proteolíticas22-24. La activación del NF-κB representa el último eslabón de la vía de señales de transducción desde la superficie celular hasta el núcleo, donde se producen las anomalías de transcripción. Hay una gran variedad de estímulos extracelulares que pueden disparar la activación del NF-κB e inducir alteraciones proliferativas de la vasculatura, incluida la aterosclerosis25-27.
Estudios clínicos muestran que los pacientes con apnea obstructiva del sueño (AOS) tienen elevadas concentraciones plasmáticas de mediadores inflamatorios, tales como citocinas (TNF-α, IL-6 e IL-18), quimiocinas (IL-8, MCP-1 proteína quimiotáctica de los monocitos-1) y moléculas de adhesión (moléculas de adhesión intercelular y vascular ICAM-1 y VCAM-1, E-selectina, P-selectina y L-selectina), así como un incremento de la expresión de contrarreceptores de moléculas de adhesión como el CD15 y CD11c en monocitos28-30. El hecho de que la AOS esté precedida o asociada a una respuesta inflamatoria, en la que muchos de los mediadores regulados por el NF-κB se hallan alterados, hace pensar que la activación de este factor nuclear puede ser uno de los mecanismos importantes en la patogenia de esta enfermedad o en sus consecuencias sistémicas. Asimismo, recien-temente se ha observado que pacientes con AOS tie-nen el NF-κB activado en monocitos31, lo que, como se verá, va a tener mucha importancia en el desarrollo de enfermedades cardiovasculares asociadas al SAHS.
Factor inducible de hipoxia (HIF-1)
Como puede observarse en la figura 6 con más detalle, el HIF-1 se expresa en todas las células nucleadas en respuesta a la hipoxia. Es un factor de transcripción que activa genes de proteínas que median la respuesta adaptativa a la reducción de la biodisponibilidad del oxígeno. Para ello participa en el metabolismo de la glucosa y energético, en la angiogenia, en la reactividad vascular y en la remodelación, en la proliferación celular y en la supervivencia célula. Entre los productos proteicos identificados de los genes diana del HIF-1 para aumentar la biodisponibilidad de oxígeno figuran, entre otros, al factor de crecimiento del endotelio vascular (VEGF), el péptido natriurético auricular, la endotelina-1, enzimas glucolíticas, la eritropoyetina y la óxido nítrico sintasa inducible27-32.
La hipoxia induce la activación de la expresión génica de HIF-1, que se inactiva rápidamente en presencia de oxígeno27. El estrés oxidativo desempeñaría un papel importante en las vías de regulación dependientes del HIF-1 tanto en condiciones normales como en condiciones patológicas33.
Actualmente muy pocos estudios clínicos han demostrado la activación directa del HIF-1 en pacientes con AOS. Todas las investigaciones se han encaminado hacia el estudio de los productos génicos expresados por el HIF-1, para demostrar indirectamente tanto la activación de éste (incremento de las concentraciones plasmáticas de VEGF34, de péptido natriurético auricular35 y de eritropoyetina36) como del precursor de la endotelina-137. La activación de HIF-1 en pacientes con AOS daría lugar a la expresión de genes antes mencionados que se relacionan con la adaptación de las células a la hipoxia.
Angiogenia y aterosclerosis
Como ya se ha mencionado, en respuesta a períodos de hipoxia/normoxia y de estrés oxidativo se activan factores de transcripción como el NF-κB y el HIF-1, entre otros. Dicha activación dará lugar a la expresión génica de factores de crecimiento, citocinas y enzimas, que, por un lado, desencadenarán una respuesta inflamatoria y, por otro, promoverán la formación de la angiogenia para incrementar la distribución de oxígeno celular y la glucólisis.
El VEGF y otras citocinas como la IL-1, IL-8 y el TNF-α intervienen en distintas etapas de la angiogenia. La IL-1 e IL-8 activan células endoteliales vasculares. La IL-1 induce la expresión de colagenasas o metaloproteinasas de la matriz, las cuales tienen un papel importante en la degradación de la matriz extracelular permitiendo la formación de nuevos vasos. Mientras que la IL-8 posee actividad angiogénica similar al TNF-α y al VEGF, no obstante la formación de nuevos vasos sanguíneos depende principalmente de las concentraciones de VEGF en tejidos, ya que su expresión esta controlada básicamente por la concentración de oxígeno34,38,39. Por otro lado, las células endoteliales sintetizan y liberan al medio extracelular factores que regulan también la angiogenia, las respuestas inflamatorias, la hemostasis, el tono y la permeabilidad vasculares.
La AOS y la angiogenia tienen como común denominador el estrés oxidativo y la inflamación implicados en la disfunción endotelial40, que finalmente puede desembocar en enfermedades cardiovasculares. Los mecanismos involucrados en el desarrollo de enfermedades cardiovasculares asociadas a la AOS pueden ser debidos a la activación de estos factores de transcripción (NF-κB, HIF-1), que predispondrían a la formación de disfunción endotelial, anomalías vasculares, lesiones en diversos tejidos y arterosclerosis.
La aterosclerosis es un daño progresivo, que se caracteriza por la acumulación de lípidos y elementos fibrosos en las arterias. Debido a las diferencias en el flujo dinámico de la sangre, hay áreas de la vasculatura más propensas a la formación de la lesión, como las ramificaciones o zonas de bifurcación41. En la figura 7 se muestran los 3 procesos que caracterizan la formación de la aterosclerosis. Ésta es el camino final de diversas enfermedades42-44. El SAHS induce arterosclerosis por una determinada serie de procesos característicos que luego van a seguir los pasos comunes de otras enfermedades que también la inducen. A modo de resumen, son los siguientes:

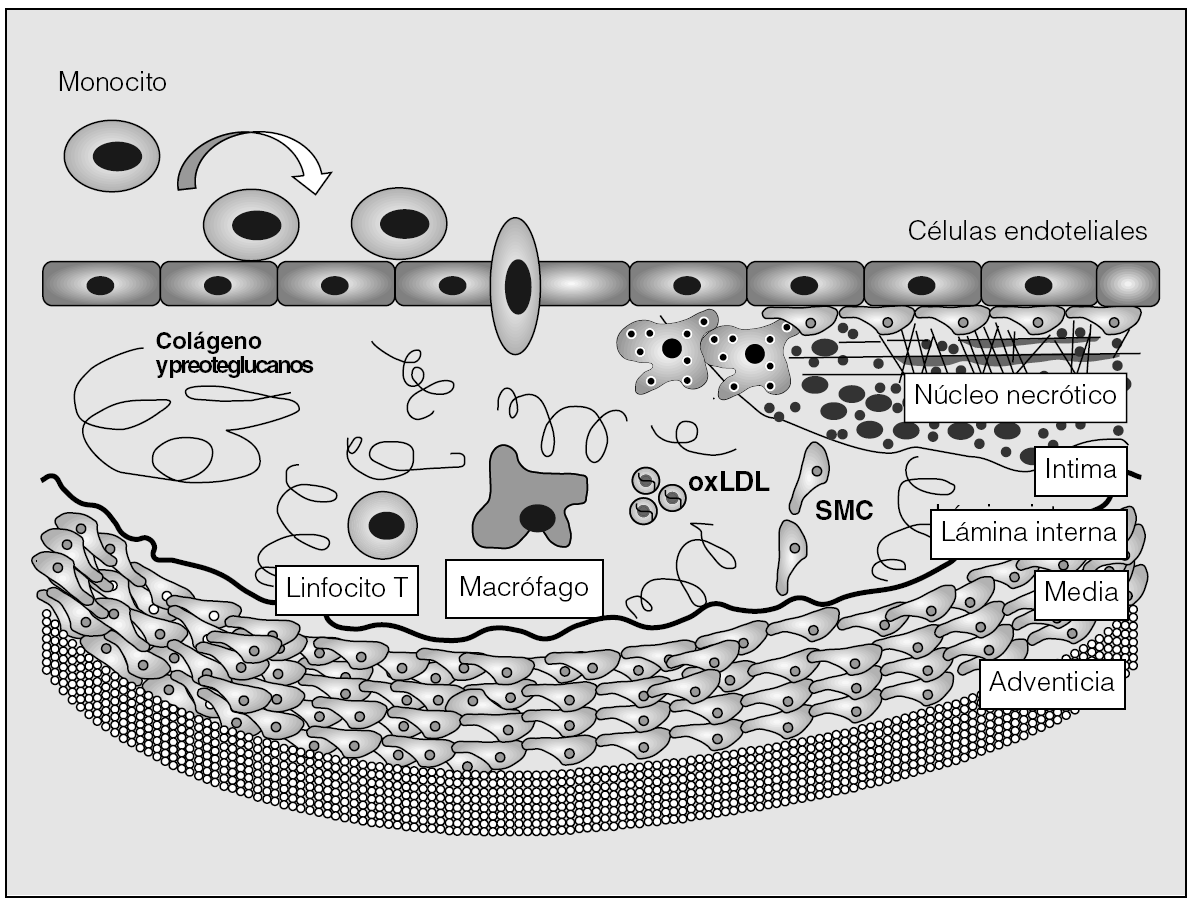
Fig. 7. Muestra el proceso de formación de la placa de ateroma. Tres son los procesos que caracterizan la formación de la aterosclerosis: 1) En la parte superior: los monocitos se adhieren al endotelio vascular, penetran la línea endotelial y entran a la íntima de la pared del vaso por diapédesis entre las células endoteliales. Este proceso requiere de un gradiente quimioatrayente como la proteína quimioatrayente de monocitos (MCP-1) o las LDL modificadas. 2) En la parte media: a) En la íntima los monocitos maduran a macrófagos, los cuales aumentan la expresión de receptores y fagocitan lipoproteínas modificadas. Los ésteres de colesterol acumulados en el citoplasma activan a los macrófagos dando lugar a las células espumosas. Estas células son características de los estadios tempranos de la aterosclerosis; b) Los macrófagos producen factores de crecimiento y citoquinas que amplifican la señal proinflamatoria; c) Los linfocitos T también migran a la íntima produciendo citoquinas proinflamatorias, que amplifican la actividad inflamatoria colaborando en la formación de la placa de ateroma. 3) En la parte inferior de la figura: se muestra la multiplicación de las células del músculo liso (SMC), las cuales migran y se acumulan en la placa, y dan una abundante matriz extracelular. Si la lesión avanza, el lumen arterial se estrecha hasta que el flujo se obstruye dando lugar a las manifestaciones clínicas.
1. La hipoxia/normoxia, como elemento diferencial, induce estrés oxidativo, activación de las vías del NF-κB y del HIF-1, entre otras, y generación de múltiples mediadores. Se produce una lesión endotelial, que altera la liberación de los mediadores que regulan el tono vascular, como el óxido nítrico.
2. Se produce el rodamiento, la adhesión y la penetración de los leucocitos en la íntima. Especialmente los monocitos que están adheridos al endotelio vascular entran en la íntima de la pared del vaso por diapédesis entre las células endoteliales. Los monocitos evolucionan a macrófagos, que fagocitan lipoproteínas modificadas.
3. Los macrófagos evolucionan a células espumosas, que son características de los estadios tempranos de la aterosclerosis. Los macrófagos, además, producen factores de crecimiento y citocinas que amplifican la señal proinflamatoria. Los linfocitos T también migran a la íntima y producen citocinas proinflamatorias que amplifican más la actividad inflamatoria y contribuyen a la formación de la placa de ateroma.
4. La placa de ateroma desarrolla canales microvasculares con función nutritiva, como resultado de la neoangiogenia, y promueve el crecimiento de la placa. En la progresión del ateroma la placa aumenta de tamaño proyectándose hacia el lumen arterial e impide el flujo de sangre45. El crecimiento de la placa de ateroma dará lugar a diferentes manifestaciones clínicas, como enfermedades cerebro y cardiovasculares asociadas a la AOS46-48.
Trabajo subvencionado por SAF2004-00684, SEPAR, FUCAP y Air-Products.
Correspondencia: Dr. J.M. Montserrat.
Unitat del Son. Servei de Pneumologia. Hospital Clínic.
Villarroel, 170. 08036 Barcelona. España.
Correo electrónico: jmmontserrat@ub.edu







