




Introducción
En los últimos años el avance en la clasificación de las enfermedades pulmonares intersticiales difusas ha permitido diagnosticar con mayor precisión cada una de las entidades y sentar una buena base para estudiar su fisiopatología. Dentro de las enfermedades pulmonares intersticiales difusas, la fibrosis pulmonar idiopática (FPI) es la que tiene mayor incidencia y peor pronóstico1,2. Además, los tratamientos actuales no mejoran la supervivencia, se desconoce la causa desencadenante y quedan muchas incógnitas en cuanto los mecanismos exactos que están implicados en la evolución1,2. Por este motivo la mayoría de los estudios básicos se han centrado en investigar su fisiopatología y cómo inhibir el proceso.
Patología básica de la respuesta pulmonar fibrótica
La hipótesis fisiopatológica actualmente aceptada en la fibrosis pulmonar es que, en individuos genéticamente predispuestos, factores externos lesionan el epitelio alveolar y se inicia un proceso reparativo alterado, que activa el depósito incontrolado de la matriz extracelular y desestructura la arquitectura pulmonar3. Esta hipótesis, basada en una amplia serie de estudios realizados durante las 2 últimas décadas, marca los campos de investigación que han de abordarse en el presente y el futuro:
1. Alteraciones genéticas que podrían predisponer a que el epitelio e intersticio pulmonares reaccionen de forma anómala ante una lesión.
2. Factores externos que activan la respuesta patológica.
3. Alteraciones celulares y moleculares, activadas tras la lesión del epitelio alveolar, que conducen a una reparación anómala del tejido.
4. Relaciones epitelio-mesenquimales alteradas que perpetúan el proceso fibrótico.
5. Factores que favorecen la progresión de la fibrosis o aceleran su curso.
El contexto fundamental que permite estudiar los mecanismos fisiopatológicos de la fibrosis pulmonar es el avance en la tecnología para el estudio molecular y la manipulación transgénica de animales para la experimentación de enfermedades complejas. Las incógnitas que promueven estos estudios tienen en común el objetivo de buscar un tratamiento efectivo de la enfermedad, entendiendo que para curar o prevenir es necesario conocer los mecanismos que la originan.
Modelos experimentales in vivo
La base para la utilización de modelos animales en la investigación de los mecanismos fibrogénicos son las similitudes estructurales, bioquímicas y moleculares entre la reacción fibrótica en humanos y animales. La experimentación en animales es el único procedimiento que permite valorar en tiempo real el efecto de las interacciones genéticas, bioquímicas y medioambientales que provocan fibrosis pulmonar. Estos modelos se utilizan para investigar un amplio abanico de eventos: a) el control de la muerte celular fisiológica (apoptosis) tanto del epitelio alveolar como de fibroblastos y miofibroblastos; b) síntesis, mecanismo de acción y regulación de mediadores fibrogénicos o antifibrogénicos. Dependiendo del mecanismo que se desea estudiar, se puede utilizar tanto la manipulación génica como la inducción externa del proceso fibrótico, y c) ensayo de nuevos fármacos antifibróticos o intervenciones que frenen la fibrogenia. No obstante, no hay ningún modelo animal que reproduzca exactamente todos los aspectos de la fibrosis pulmonar en humanos. Por este motivo la utilidad y elección del modelo animal a utilizar estarán condicionadas por las características del evento fibrótico determinado que se quiere estudiar, las hipótesis y los objetivos de la investigación, así como las consideraciones logísticas experimentales de las que se dispone.
Los animales de experimentación más utilizados son el ratón y la rata, por su fácil manipulación y el menor coste, aunque se ha estudiado un amplio grupo de animales. Los métodos convencionales para inducir reacciones pulmonares fibróticas incluyen la instilación directa de agentes fibrogénicos y la exposición a irradiación torácica4-6 (tabla I). Entre los agentes fibrogénicos, el más utilizado es la bleomicina, un potente agente antineoplásico que actúa mediante un efecto oxidante, escindiendo el ADN y aumentando los mediadores fibrogénicos. Se puede administrar de forma endotraqueal, intraperitoneal o intravenosa, y raramente por vía intramuscular o subcutánea. Las primeras observaciones del efecto profibrótico de este agente antineoplásico fueron en perros7. Posteriormente se estandarizó la instilación endotraqueal en dosis única en ratas y ratones, hasta determinar la dosis requerida para provocar cambios pulmonares fibróticos uniformes, muy similares a los observados en la FPI8,9. Las lesiones inducidas por este agente incluyen inflamación parcheada y variable, lesión epitelial con hiperplasia reactiva y apoptosis, alteración de la membrana basal, fibrosis de distribución heterogénea (intraalveolar, subpleural o bronquiolocéntrica), así como focos de células mesenquimales en forma de huso de apariencia similar a los focos de miofibroblastos observados en la neumonía intersticial usual10. En los últimos años el modelo de bleomicina se ha utilizado para: a) estudiar los mecanismos de acción de factores de crecimiento (como el factor transformador del crecimiento beta, la angiotensina II y la endotelina-1) y de diversas citocinas, así como el efecto terapéutico de su inhibición11-17; b) valorar la modulación transgénica en la respuesta fibrótica e identificar posibles loci reguladores en la predisposición genética a la fibrosis pulmonar16,18, y c) estudiar la alteración celular epitelial, fibroblástica y de la matriz extracelular19-21. Aunque el modelo de bleomicina instilada es fácilmente reproducible, cabe recordar que es un modelo de lesión de rápida instauración y duración determinada. Para estudiar eventos iniciales, donde predomina la inflamación, se sacrifica a los animales a partir del tercer al séptimo días. Asimismo, si lo que se pretende es estudiar la fase fibrótica de la lesión, lo ideal es que los animales se evalúen las semanas 2 y 3, dado que el daño provocado revierte espontáneamente a partir de la tercera semana9. Los modelos de administración continua de bleomicina que se han ensayado hasta la actualidad no acaban de reproducir con exactitud la cronicidad de la fibrosis pulmonar22.

La inducción de fibrosis con amiodarona es otro modelo bien estandarizado en rata, ratón y hámster. Se utiliza para el estudio de su toxicidad y, administrada por vía endotraqueal y oral, se ha observado que el efecto profibrótico deriva tanto de este compuesto como de su metabolito, la desetilamiodarona23-25. La inhalación de partículas como el asbesto o sílice induce lesiones pulmonares similares en humanos y roedores: provoca fibrosis de forma progresiva, acompañada de reacción inflamatoria granulomatosa. Estos modelos son especialmente útiles para el estudio de macrófagos y otros fagocitos26,27. Otros elementos pueden inducir fibrosis en animales, como la inhalación de cobalto y cloruro de cadmio, paraquat o diquat vía sistémica, isocianatos y nitrosoureas, o pentóxido de vanadio, aunque su uso para el estudio de este proceso no ha sido estandarizado. La irradiación como inductor de fibrosis es también un sistema muy utilizado. Las dosis empleadas son variables y los efectos fibróticos pueden persistir varios meses tras la radiación28-30. El inconveniente principal es el aparataje, que comporta la necesidad de grandes espacios, así como una rigurosa utilización y mantenimiento del sistema.
Los avances en la manipulación genética de animales de experimentación, en particular de los ratones, han permitido estudiar patrones de susceptibilidad a la fibrosis pulmonar, tales como alteraciones en la expresión de citocinas y factores de crecimiento, de componentes de la matriz extracelular o de otros factores específicos que pueden estar implicados en este proceso (enzimas, proteínas y otros componentes intersticiales y del epitelio alveolar). Los animales transgénicos portan un fragmento de ADN exógeno en su genoma, lo cual permite el estudio del patrón de expresión de ese gen y las consecuencias biológicas de la sobreexpresión de la proteína exógena codificada por el transgén en tejidos específicos. La mutagenia de un gen ha permitido generar cepas de ratones que carecen de proteínas determinadas, con lo que se desarrollan modelos específicos de pérdida de función; es lo que se conoce como animales knock-out. Cuando el gen normal se sustituye por otro alterado, con mutaciones específicas, recibe el nombre de knock-in. Actualmente es posible utilizar ratones con deleciones completas de un gen determinado para comprobar el efecto de la ausencia completa de su producto26,31-35, estudiar los cambios en la expresión génica que originan diferentes inductores36,37, o bien provocar diferentes mutaciones genéticas para evaluar determinadas vías terapéuticas38. Para decidir qué genes estudiar, es útil investigar previamente cómo influye la inducción del proceso fibrótico en el cambio de la expresión genética. Este análisis se ha visto facilitado por la aparición de la técnica de micromatrices (microarrays)39, que consiste en el análisis de la expresión simultánea de miles de genes mediante la tecnología de organización de micromatrices de ADN. Con este método es posible determinar la alteración del patrón transcripcional de múltiples genes en un mismo tejido tras la inducción de fibrosis o manipulación genética experimental previa40-43. En los últimos años se han cruzado ratones transgénicos para obtener modificaciones génicas combinadas que permiten evaluar varios factores implicados en el proceso de fibrogenia44. Este método condiciona un mayor ajuste a la realidad, dado que en los últimos años se ha postulado que la FPI es una enfermedad poligénica; es decir, el individuo afectado presentaría más de una alteración genética45. El inconveniente básico de trabajar con ratones transgénicos es el elevado coste y la dificultad para obtener algunos modelos, dado que son pocos los laboratorios que trabajan en este campo. Probablemente la mayor accesibilidad al uso de estos animales sea una de las mejoras que se produzcan en los próximos años.
Sin embargo, se sabe que la historia natural de la fibrosis pulmonar en humanos difiere en varios aspectos de la inducida en animales. Por una parte, no hay ningún modelo que consiga una reproducción precisa de la cronicidad o progresión de la enfermedad, probablemente por el desconocimiento de los factores que provocan en el pulmón humano que un evento reparativo patológico lleve a la perpetuación de la fibrosis46. Además, en la mayoría de los modelos animales los cambios producidos por una inducción inicial tienden a regresar, lo cual no ocurre en el proceso humano. Por estos motivos, para extrapolar a la clínica los resultados obtenidos en un modelo animal será primordial su correcta y estricta interpretación. La tendencia a experimentar con fenotipos que reproduzcan lo máximo posible el proceso en humanos es la base para mejorar los modelos animales en el estudio de la fibrosis pulmonar en un futuro próximo.
Modelos experimentales in vitro
Los modelos experimentales in vitro comprenden una amplia serie de métodos cuyo objetivo es evaluar una respuesta celular o molecular determinada. Los estudios morfológicos aportan información relevante sobre el desarrollo y la progresión de la fibrosis. Sin embargo, estos modelos sólo permiten evaluar la expresión de una determinada molécula y comparar sus características respecto al tejido sano. Para evaluar si estos hallazgos tienen una implicación activa en el proceso fibrótico y determinar su papel, es indispensable trabajar con modelos que permitan la funcionalidad del tejido o la actividad de las células que intervienen. Suelen utilizarse explantes de tejido pulmonar o cultivos de células inflamatorias y mesenquimales, obtenidas de modelos animales y de pacientes con fibrosis pulmonar.
Los explantes pulmonares permiten investigar, en diferentes condiciones (basalmente, después de añadir factores inductores o un fármaco determinado), la expresión de una molécula, su síntesis y su tipificación genética. Habitualmente estos estudios se llevan a cabo durante pocas horas o días, dado que el explante se degenera fácilmente47,48.
El cultivo de células a partir de tejido pulmonar permite el estudio de su proliferación, fenotipo y receptores expresados, así como de los productos que sintetiza. En las placas de cultivo (fig. 1) el fibroblasto es una célula que crece y se adhiere con facilidad. El método óptimo para su obtención es aprovechando un trozo de la pieza de biopsia quirúrgica del paciente para el diagnóstico de la enfermedad intersticial. Los sujetos que pueden considerarse estrictamente controles son aquellos en los que la pieza histológica se cataloga de normal, cuyo patrón funcional respiratorio carece de alteraciones y que no presentan enfermedad inflamatoria o infecciosa concomitante. La utilización de cultivos de fibroblastos49,50 ha permitido valorar las características de los fibroblastos-miofibroblastos en la FPI y otras enfermedades intersticiales, su resistencia a la apoptosis y la síntesis de moléculas profibróticas, así como los componentes de la matriz extracelular42,49-53. Otra célula elemental para investigar es la célula alveolar epitelial tipo II. Estas células son difíciles de obtener a partir del tejido humano, por lo que se utilizan líneas de células epiteliales normales derivadas de la manipulación biológica, como la línea A54916,54-57. También se pueden obtener células epiteliales patológicas de los modelos animales de fibrosis mediante el lavado broncoalveolar o digestión por colagenasa de la pieza pulmonar una vez sacrificado el animal55,58. Los macrófagos intervienen asimismo en el proceso fibrótico y su estudio se ve facilitado por su fácil obtención mediante el lavado broncoalveolar de pacientes con fibrosis pulmonar o de animales con fibrosis pulmonar inducida, aunque en ratones otra forma de obtenerlos es la vía peritoneal44. Los principales inconvenientes son que el estudio funcional de los macrófagos se limita a 24 h y que su cultivo requiere medidas cualitativas estrictas para optimizar la pureza celular, es decir, evitar el crecimiento de otras células como fibroblastos o eritrocitos59,60.
Fig. 1. Placas de cultivo. Las más utilizadas son de 6; 12; 48, y 96 pocillos.
Otro método que permite una aproximación más exacta a los fenómenos que podrían darse en el tejido pulmonar de los pacientes con fibrosis pulmonar son los cocultivos celulares. Mediante placas especiales que permiten confrontar 2 tipos celulares en proliferación se puede estudiar el efecto que tienen uno sobre el otro o los mediadores que secretan e intervienen en la fibrogenia. La interacción entre los tipos celulares se hace posible gracias a una fina membrana que separa los 2 compartimentos (fig. 2). Este método resulta ideal para evaluar la interacción entre las células epiteliales alveolares y los fibroblastos-miofibroblastos61,62, así como la relación macrófago-célula epitelial-fibroblasto63, contexto que más se aproxima a los fenómenos que se producen en el pulmón de los pacientes fibróticos. Este campo más complejo y novedoso está progresando dada la variedad de opciones que permite, por lo que podría mejorar el diseño de los experimentos en los próximos años.

Fig. 2. Placas de cocultivos celulares. Son placas con una membrana que permite separar los diferentes tipos celulares o medios compartimentales.
Avance en el conocimiento de la fibrosis pulmonar a través de los modelos experimentales. Implicaciones en la práctica clínica
En la última década, los modelos experimentales de fibrosis pulmonar han aportado información muy relevante sobre su fisiopatología y han servido para investigar posibles fármacos que frenen esta enfermedad. Gracias a ellos ha quedado cimentada la nueva hipótesis sobre la fibrogenia pulmonar, en la que la célula epitelial y su apoptosis acelerada tienen un papel principal junto con los fibroblastos-miofibroblastos3,52,54,58,64 y la implicación de mediadores profibróticos46-48,50,65. Se ha comprobado que tras la lesión de la célula epitelial alveolar, inducida por factores exógenos, el proceso reparativo que se inicia es anómalo: mayor apoptosis del epitelio y permeabilidad de la membrana basal, e incremento, en el espacio alveolar e intersticio, de los mediadores profibróticos, que activan la proliferación incontrolada de fibroblastos, su cambio fenotípico a miofibroblastos y el ambiente propicio para el depósito excesivo de colágeno. En cuanto a la valoración del papel de la respuesta celular inflamatoria en la fibrosis pulmonar, aunque muchos han sido los intentos de aclarar su función, la variabilidad de los resultados obtenidos hasta el momento ha hecho imposible llegar a una conclusión definitiva. Las células inflamatorias, que sí participan en el resto de enfermedades pulmonares intersticiales, es probable que no tengan un papel primordial en la FPI, aunque se desconoce la función exacta de su incremento en el proceso fibrótico66,67. En este sentido, otro fenómeno aún sin esclarecer es el papel de la vascularización en la fibrogenia. Mientras que algunos estudios experimentales proponen que esta enfermedad se acompaña de una disminución de la vascularización, otros abogan por su aumento como parte del proceso de fibrogenia68,69. Una hipótesis es que en las zonas más afectadas por la panalización la vascularización disminuye, en tanto que en las zonas con más actividad inflamatoria existe un aumento de la permeabilidad vascular70-72. Otro aspecto que sigue siendo trascendental para la investigación es encontrar qué factor externo es inductor o iniciador del daño celular epitelial que activa todo el proceso. Desde la inhalación de algunas sustancias73 hasta la relevancia de algunos virus60,74 o la implicación del reflujo gastroesofágico75, han sido diversas las hipótesis postuladas, pero sin conclusiones evidentes. Asimismo, esta incógnita se acompaña del complejo componente endógeno humano: la predisposición genética. Se sabe que existe un tipo de fibrosis pulmonar hereditaria76,77, además de cierta agregación familiar en pacientes afectados de diversas enfermedades intersticiales78,79. Los estudios genéticos realizados se centran en el análisis de mutaciones de citocinas, factores de crecimiento, componentes del complejo principal de histocompatibilidad e integrantes de la matriz extracelular y epitelio45,80-84.
Tras observar el escaso beneficio de la pauta de tratamiento clásica para la FPI (glucocorticoides más inmunodepresores), han sido múltiples los esfuerzos para encaminar las investigaciones hacia tratamientos antifibróticos efectivos85. En este sentido, varios modelos experimentales han servido para valorar inicialmente fármacos antifibróticos y han abierto expectativas para su aplicación en humanos. En la actualidad algunos de ellos son objeto de ensayos clínicos85, como el interferón γ1b, la pirfenidona, la N-acetilcisteína y el bosentán. Por lo tanto, los modelos experimentales de fibrosis pulmonar no sólo aclaran puntos clave de la fisiopatología que han permitido avanzar en el conocimiento de su evolución, sino que también son una herramienta indispensable para evaluar la seguridad y acción de nuevos fármacos antifibróticos (fig. 3).
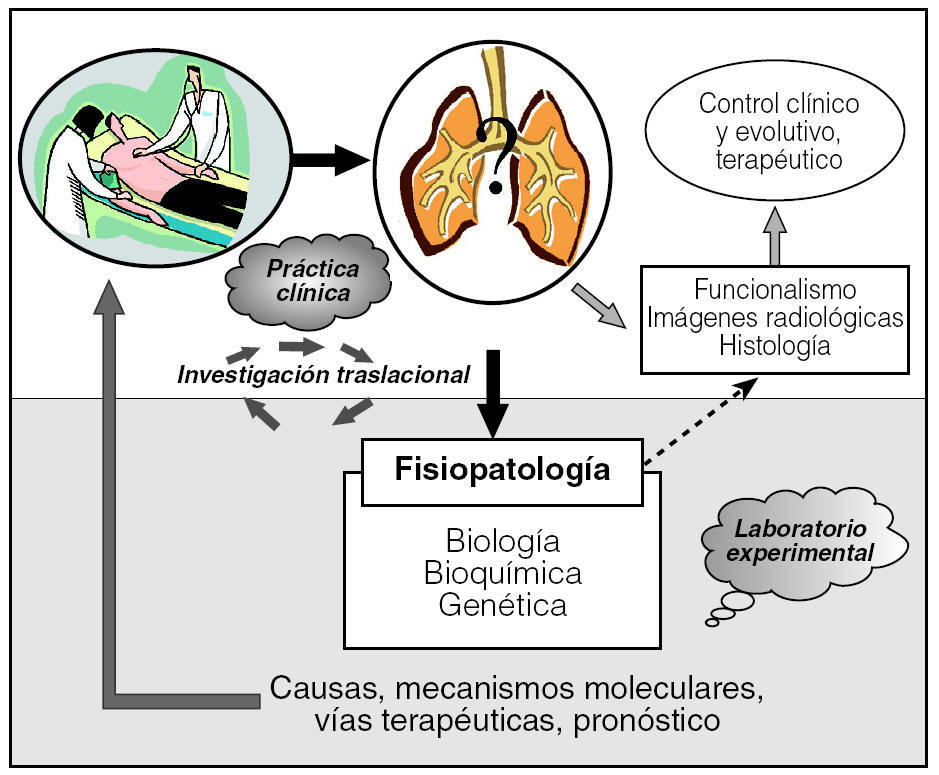
Fig. 3. Investigación traslacional: los estudios experimentales son una herramienta indispensable para la investigación básica de las enfermedades respiratorias.
En conclusión, el estudio básico experimental de la fibrosis pulmonar, así como del resto de enfermedades respiratorias, nace de las mil preguntas planteadas en la práctica clínica y experimental, intenta desglosarlas en busca de respuestas específicas y, finalmente, aporta unos resultados que serán parte del conocimiento y, muy probablemente, el desencadenante de nuevas preguntas o hipótesis.
Correspondencia: Dra. M. Molina-Molina.
Unidad de Endoscopia Respiratoria. Servicio de Neumología. Hospital Clínico.
Villarroel, 170. Barcelona. España.
Correo electrónico: mariamolinamolina@hotmail.com
Recibido: 12-9-2006; aceptado para su publicación: 19-9-2006.







