


La gran mayoría de las enfermedades respiratorias son consideradas patologías complejas puesto que su susceptibilidad o desenlace están influidos por la interacción entre factores dependientes del huésped (genéticos, comorbilidad, edad, etc.) y del ambiente (exposición a microorganismos y alérgenos, tratamiento administrado, etc.).
El enfoque reduccionista ha sido muy importante para la comprensión de los diversos componentes de un sistema. La biología o medicina de sistemas es una aproximación complementaria cuyo objetivo es el análisis de las interacciones entre los componentes dentro de un nivel de organización (genoma, transcriptoma, proteoma) y posteriormente entre los distintos niveles.
Las actuales aplicaciones de la medicina de sistemas incluyen la interpretación de la patogénesis y fisiopatología de las enfermedades, el descubrimiento de biomarcadores, el diseño de nuevas estrategias terapéuticas y la elaboración de modelos computacionales para los distintos procesos biológicos.
En la presente revisión se exponen las principales nociones sobre la teoría que subyace a la medicina de sistemas así como sus aplicaciones en algunos procesos biológicos del ser humano.
Most respiratory diseases are considered complex diseases as their susceptibility and outcomes are determined by the interaction between host-dependent factors (genetic factors, comorbidities, etc.) and environmental factors (exposure to microorganisms or allergens, treatments received, etc.)
The reductionist approach in the study of diseases has been of fundamental importance for the understanding of the different components of a system. Systems biology or systems medicine is a complementary approach aimed at analyzing the interactions between the different components within one organizational level (genome, transcriptome, proteome), and then between the different levels.
Systems medicine is currently used for the interpretation and understanding of the pathogenesis and pathophysiology of different diseases, biomarker discovery, design of innovative therapeutic targets, and the drawing up of computational models for different biological processes.
In this review we discuss the most relevant concepts of the theory underlying systems medicine, as well as its applications in the various biological processes in humans.
La gran mayoría de las enfermedades respiratorias son consideradas actualmente como patologías complejas, puesto que su susceptibilidad o desenlace están influidos por la interacción entre factores dependientes del huésped (genéticos, comorbilidad, edad, etc.) y del ambiente (exposición a microorganismos y alérgenos, tratamiento administrado, etc.).
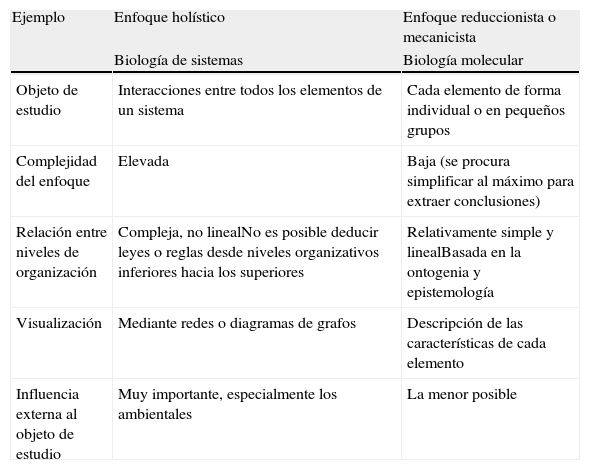
La aparición de técnicas nuevas y sofisticadas en la primera mitad del siglo xx permitió comenzar a conocer el funcionamiento, la estructura y los detalles de las partes de cada sistema o proceso biológico. Fue el auge del enfoque reduccionista o mecanicista.
A inicios del presente siglo, y estimulado nuevamente por el desarrollo tecnológico capaz de producir datos experimentales e in silico de forma masiva, rápida y a un relativo bajo coste, se generó un cambio en el paradigma científico desplazando el foco de interés desde el estudio de las partes al estudio de las interacciones que existen dentro de un sistema. Una enfermedad específica puede ser considerada como un sistema.
La biología de sistemas, por tanto, puede ser definida como el área de estudio que se ocupa del análisis de las interacciones complejas dentro de un sistema con distintas escalas de organización biológica, desde moléculas a células, órganos, individuos, sociedad y ecosistema. La biología de sistemas se caracteriza por buscar una descripción cuantitativa de los procesos biológicos, que incluyen múltiples niveles (genoma, transcriptoma, proteoma, metaboloma, etc.) y distintas escalas de tiempo, valiéndose de datos generados generalmente por tecnología de alta eficiencia (high throughput), algoritmos matemáticos y modelos computacionales1. Cuando el concepto de biología de sistemas se aplica al área de las ciencias de la salud se denomina medicina de sistemas. Biología de sistemas y medicina de sistemas son conceptos prácticamente idénticos, con la excepción de que el primero es general y el segundo está particularmente enfocado a la medicina. Otra característica particular de la medicina de sistemas es la frecuente utilización de los diagramas de grafos, que tienen reglas específicas (analizadas en el presente artículo) al tiempo que constituyen una forma sencilla de comprender y visualizar los resultados.
La medicina de sistemas es un área del conocimiento que se encuentra en desarrollo exponencial y que tiene claras implicaciones clínicas. Algunas de sus aplicaciones incluyen el desarrollo de modelos fisiológicos y fisiopatológicos, el descubrimiento de nuevos fármacos, y el desarrollo de pruebas diagnósticas y de nuevos biomarcadores (tabla 1).
Comparación entre los enfoques reduccionista y holístico
| Ejemplo | Enfoque holístico | Enfoque reduccionista o mecanicista |
| Biología de sistemas | Biología molecular | |
| Objeto de estudio | Interacciones entre todos los elementos de un sistema | Cada elemento de forma individual o en pequeños grupos |
| Complejidad del enfoque | Elevada | Baja (se procura simplificar al máximo para extraer conclusiones) |
| Relación entre niveles de organización | Compleja, no linealNo es posible deducir leyes o reglas desde niveles organizativos inferiores hacia los superiores | Relativamente simple y linealBasada en la ontogenia y epistemología |
| Visualización | Mediante redes o diagramas de grafos | Descripción de las características de cada elemento |
| Influencia externa al objeto de estudio | Muy importante, especialmente los ambientales | La menor posible |
La presente revisión está dirigida a médicos clínicos y pretende transmitir de una manera sencilla las nociones básicas de la medicina de sistemas así como algunas de sus aplicaciones.
Medicina de sistemasEl enfoque tradicional reduccionista se centra en el análisis de moléculas o procesos específicos desde el punto de vista individual basándose en 2 tipos de conceptos:
- (a)
Conceptos ontológicos: todas las cosas están constituidas por un limitado grupo de elementos materiales primitivos e indivisible. Conocer la interacción entre estos compuestos básicos es suficiente para explicar todos los fenómenos complejos.
- (b)
Conceptos epistemológicos: las leyes y teorías en una determinada área y nivel de organización se originan desde niveles organizativos inferiores (más básicos)2.
La biología molecular clásica utiliza de forma prácticamente privativa el enfoque reduccionista, puesto que se basa principalmente en la caracterización de moléculas o genes y en explicar los procesos biológicos mediante combinaciones de interacciones y propiedades entre sus componentes.
Sin embargo, el estudio del genoma, el metabolismo celular y las interacciones entre proteínas puede ser abordado mediante técnicas e interpretaciones globales (aproximación holística). El enfoque holístico basa su potencia en la identificación y análisis de las interacciones existentes entre los distintos elementos (nodos) de una red. Estas interacciones son complejas e incluyen múltiples niveles de organización (ADN, ARN, proteínas y ambiente, por ejemplo); no son lineales (no existe una relación directamente proporcional y fácilmente deducible entre un nivel de organización y otro); y son redundantes y con múltiples circuitos de retroalimentación. En este enfoque holístico los conceptos de ontogenia y epistemología no son aplicables, pues a un elemento no se le puede asignar una función exclusiva, como tampoco es posible deducir las reglas o leyes de un nivel en función de otro nivel organizativo.
La aplicación del concepto de biología de sistemas a problemas médicos específicos ha dado origen a la medicina de sistemas, que permite establecer nuevas asociaciones entre funciones biológicas y enfermedades o condiciones humanas especiales. Por ejemplo, la sepsis grave está provocada por un daño infeccioso (bacterias, virus, parásitos), en un determinado momento (en la comunidad o durante la estancia en ella) y en un huésped con determinadas características (comorbilidades, estado nutricional, estado inmunitario, portador de determinados polimorfismos genéticos). La respuesta que se genera (alteraciones del transcriptoma o del proteoma) está claramente influida por los factores previamente descritos y por el tratamiento administrado. El objetivo de la medicina de sistemas es analizar todos estos factores de una manera holística e integrada, priorizando la articulación entre los distintos niveles de organización sobre el funcionamiento de cada elemento en particular.
Diagramas de grafos y redes biológicasLos diagramas de grafos aplicados a procesos biológicos son denominados redes biológicas y constituyen una forma de visualizar la información de una forma fácil y compresible. Un grafo es un conjunto de objetos llamados vértices o nodos conectados entre sí por enlaces. Posiblemente los grafos más conocidos posiblemente sean los mapas de las conexiones aéreas. Los nodos son los elementos de interés, por ejemplo, proteínas, genes, metabolitos o, en el ejemplo antes mencionado, los aeropuertos, mientras que los enlaces son las relaciones existentes entre los nodos y que se corresponderían a las rutas de los aviones entre 2 aeropuertos. Un grafo, por tanto, está definido por un conjunto V de nodos (por ejemplo, aeropuertos) y E de enlaces (por ejemplo, rutas), recordando que cada enlace tiene 2 nodos (el avión debe salir desde y llegar a un aeropuerto). A cada enlace se le puede asignar un peso, una dirección y un tipo.
Los grafos son empleados en medicina principalmente con 3 objetivos:
- (a)
Representar el conocimiento de, por ejemplo, rutas metabólicas, señales de transducción o expresión génica.
- (b)
Cuantificar y visualizar los datos producidos mediante experimentos y técnicas de alta eficiencia (high-throughput), por ejemplo los datos generados mediante microarrays o espectroscopia de masas.
- (c)
Como modelos estadísticos. Por ejemplo, para estimar qué elementos participan en la formación de una proteína partiendo de un volumen de datos determinados. Los grafos permiten elaborar distintos modelos, y luego elegir cuál de ellos se adapta mejor a la realidad observada experimentalmente.
Los parámetros topológicos de una red biológica son las características propias de ella. Su conocimiento es fundamental para comprender su arquitectura y funcionamiento. Las características más importantes son3 (tabla 2):
- (a)
Grado (degree). Es el número de conexiones que tiene un nodo. Se representa con la letra k. En digrafos (tabla 2) es posible discriminar entre un grado de entrada (in-degree) y uno de salida (out-degree). En general, cuanto mayor es el grado de un nodo, mayor es su importancia en la red (los hubs se destacan por un grado superior al resto de los nodos).
- (b)
Distancia (distance). Es el camino mínimo entre 2 nodos.
- (c)
Diámetro (diameter). Es la máxima distancia entre 2 nodos.
- (d)
Coeficiente de agrupamiento (clustering coefficient). Es la proporción entre el número de conexiones de los vecinos de un nodo y el número de conexiones máximas que podrían tener. El cálculo se realiza para cada nodo en particular (fig. 1).
 y distinto coeficiente de agrupamiento: la red A es de 2/5 (2 conexiones de las 5 potencialmente posibles); la red B es de 1 (todas las conexiones posibles están presentes). Panel superior-derecha. Se representan los 4 tipos principales de redes (diagramas de grafos). Los hexagonales representan los nodos y las líneas las conexiones. 1a, red aleatoria: las conexiones entre los nodos se establecen al azar. 1b, red regular: todos los nodos tiene el mismo número de conexiones (en el ejemplo es 2). 1c, red de mundo pequeño: es similar a la red regular pero existen «caminos» más cortos que se representan con las líneas azules. 1d, red libre de escala: existen nodos que son más importantes pues reciben más conexiones (en el ejemplo el amarillo es el nodo más importante, y los naranja tienen una importancia un poco menor) y se establecen subgrupos independientes. Panel inferior-izquierda. Se representan los sistemas biológicos. Son redes libres de escala con características particulares, en especial la presencia de módulos relativamente independientes y una organización jerárquica. En el esquema se representan el origen de la red en el módulo «amarillo» central, que es jerárquicamente el superior y por tanto el de mayor número de conexiones. Otros 3 (celeste, verde y azul) corresponden a módulos de menor jerarquía, que podrían vincularse a determinadas funciones. Por último, el módulo marrón, que es el de menor jerarquía. La mayoría de las especies conservan los módulos de mayor jerarquía (en este caso el amarillo), pues están vinculados a procesos esenciales para la vida. Contrariamente, los módulos de menor jerarquía, por ejemplo el marrón, son los que explican la diferencia entre las especies y los que permiten la adaptación a circunstancias o ambientes particulares. En verde se representa un módulo cuyo encendido o apagado depende de un patrón temporal o ambiental. Panel inferior-derecha. Se representa la estructura en forma de «pajarita» de las redes metabólicas. Están conformadas por 4 partes: (a) bloques rojos, nodos de sustratos; (b) bloques verdes, nodos de productos; (c) bloques azules, nodos independientes; y (d) el hexágono central, correspondiente al giant strong component, que incluye las vías metabólicas más importantes, como la glucólisis o el ciclo de Krebs. Los colores indicados se aprecian como escala de grises. Puede apreciarse esta figura a todo color únicamente en la versión electrónica del artículo.") Figura 1.
Figura 1.Panel superior-izquierda. Se representan 2 redes biológicas con su correspondiente nodo central (amarillo) y distinto coeficiente de agrupamiento: la red A es de 2/5 (2 conexiones de las 5 potencialmente posibles); la red B es de 1 (todas las conexiones posibles están presentes).
Panel superior-derecha. Se representan los 4 tipos principales de redes (diagramas de grafos). Los hexagonales representan los nodos y las líneas las conexiones. 1a, red aleatoria: las conexiones entre los nodos se establecen al azar. 1b, red regular: todos los nodos tiene el mismo número de conexiones (en el ejemplo es 2). 1c, red de mundo pequeño: es similar a la red regular pero existen «caminos» más cortos que se representan con las líneas azules. 1d, red libre de escala: existen nodos que son más importantes pues reciben más conexiones (en el ejemplo el amarillo es el nodo más importante, y los naranja tienen una importancia un poco menor) y se establecen subgrupos independientes.
Panel inferior-izquierda. Se representan los sistemas biológicos. Son redes libres de escala con características particulares, en especial la presencia de módulos relativamente independientes y una organización jerárquica. En el esquema se representan el origen de la red en el módulo «amarillo» central, que es jerárquicamente el superior y por tanto el de mayor número de conexiones. Otros 3 (celeste, verde y azul) corresponden a módulos de menor jerarquía, que podrían vincularse a determinadas funciones. Por último, el módulo marrón, que es el de menor jerarquía. La mayoría de las especies conservan los módulos de mayor jerarquía (en este caso el amarillo), pues están vinculados a procesos esenciales para la vida. Contrariamente, los módulos de menor jerarquía, por ejemplo el marrón, son los que explican la diferencia entre las especies y los que permiten la adaptación a circunstancias o ambientes particulares. En verde se representa un módulo cuyo encendido o apagado depende de un patrón temporal o ambiental.
Panel inferior-derecha. Se representa la estructura en forma de «pajarita» de las redes metabólicas. Están conformadas por 4 partes: (a) bloques rojos, nodos de sustratos; (b) bloques verdes, nodos de productos; (c) bloques azules, nodos independientes; y (d) el hexágono central, correspondiente al giant strong component, que incluye las vías metabólicas más importantes, como la glucólisis o el ciclo de Krebs. Los colores indicados se aprecian como escala de grises. Puede apreciarse esta figura a todo color únicamente en la versión electrónica del artículo.
- (e)
Intermediación (betweenness). Es la frecuencia con que aparece un nodo en el camino más corto que conecta otros 2 nodos. Es una estimación del tráfico que existe en el mencionado nodo.
 y distinto coeficiente de agrupamiento: la red A es de 2/5 (2 conexiones de las 5 potencialmente posibles); la red B es de 1 (todas las conexiones posibles están presentes). Panel superior-derecha. Se representan los 4 tipos principales de redes (diagramas de grafos). Los hexagonales representan los nodos y las líneas las conexiones. 1a, red aleatoria: las conexiones entre los nodos se establecen al azar. 1b, red regular: todos los nodos tiene el mismo número de conexiones (en el ejemplo es 2). 1c, red de mundo pequeño: es similar a la red regular pero existen «caminos» más cortos que se representan con las líneas azules. 1d, red libre de escala: existen nodos que son más importantes pues reciben más conexiones (en el ejemplo el amarillo es el nodo más importante, y los naranja tienen una importancia un poco menor) y se establecen subgrupos independientes. Panel inferior-izquierda. Se representan los sistemas biológicos. Son redes libres de escala con características particulares, en especial la presencia de módulos relativamente independientes y una organización jerárquica. En el esquema se representan el origen de la red en el módulo «amarillo» central, que es jerárquicamente el superior y por tanto el de mayor número de conexiones. Otros 3 (celeste, verde y azul) corresponden a módulos de menor jerarquía, que podrían vincularse a determinadas funciones. Por último, el módulo marrón, que es el de menor jerarquía. La mayoría de las especies conservan los módulos de mayor jerarquía (en este caso el amarillo), pues están vinculados a procesos esenciales para la vida. Contrariamente, los módulos de menor jerarquía, por ejemplo el marrón, son los que explican la diferencia entre las especies y los que permiten la adaptación a circunstancias o ambientes particulares. En verde se representa un módulo cuyo encendido o apagado depende de un patrón temporal o ambiental. Panel inferior-derecha. Se representa la estructura en forma de «pajarita» de las redes metabólicas. Están conformadas por 4 partes: (a) bloques rojos, nodos de sustratos; (b) bloques verdes, nodos de productos; (c) bloques azules, nodos independientes; y (d) el hexágono central, correspondiente al giant strong component, que incluye las vías metabólicas más importantes, como la glucólisis o el ciclo de Krebs. Los colores indicados se aprecian como escala de grises. Puede apreciarse esta figura a todo color únicamente en la versión electrónica del artículo.")
Nomenclatura referente a los diagramas de grafos o redes
| Nodos vecinos o adyacentes: son los 2 nodos que están conectados por una rama o arco |
| Enlaces vecinos o adyacentes: son los enlaces que están conectados por un nodo |
| Grafo simple: un grafo sin bucles ni ramas paralelas |
| Multigrafo: grafo con ramas paralelas pero sin bucles |
| Digrafos o grafos dirigidos: grafos cuyos arcos están dirigidos |
| Grafo regular: todos los nodos tienen el mismo grado |
| Grado de un nodo: el número de vecinos que tiene dicho nodo. Si existen arcos dirigidos puede calcularse el grado de entrada y de salida de dicho nodo |
| Grafo conexo: para cada par de nodos del grafo existe al menos un camino que los une |
| Bucle: rama que empieza y termina en el mismo nodo |
| Seudografo: grafo que contiene bucles |
| Bosque: grafo sin ciclos (acíclico) |
| Ramas paralelas: 2 ramas que conectan el mismo par de vértices |
| Paseo de un nodo A a un nodo Z: la secuencia alternativa de nodos y arcos que se deben recorrer para llegar desde el vértice x al y |
| Rastro: es un recorrido en el cual no se repiten las ramas |
| Camino: recorrido en el cual todos los nodos son distintos |
| Longitud: el número de ramas de un recorrido cuando ningún enlace tiene un peso específico (si algún enlace tiene un peso específico debe realizarse una corrección) |
| Camino mínimo o distancia entre el nodo A y el Z: la mínima longitud (número de ramas) que es necesario recorrer entre A y Z o viceversa |
| Paseo cerrado: recorrido o circuito en el cual no se repiten las ramas |
| Ciclo: circuito en el que no se repiten los nodos |
| Punto de articulación: un nodo que desconecta un grafo conexo |
| Corte: conjunto de ramas que desconecta un grafo conexo; si un corte está compuesto por una única rama, se denomina puente |
| Corte mínimo: mínimo número de ramas que, si son eliminadas, desconectan el grafo |
| Clique: subconjunto de un grafo no dirigido X en el cual todos los vértices están unidos por un enlace. Si se considera exclusivamente al clique, este es un grafo completo |
| Tamaño de un clique: número de vértices que contiene |
En función de sus características topológicas las redes biológicas más importantes pueden clasificarse en (fig. 1):
- (a)
Aleatorias. Las conexiones entre los nodos se producen al azar (los grados de los nodos siguen una distribución de Poisson), es decir, existe igual probabilidad de conectarse a un nodo que a otro y, por tanto, no es predecible en qué nodo se producirá la conexión.
- (b)
Libres de escala. Se trata de redes donde existe un pequeño grupo de nodos, denominados cubos (hubs), que están altamente conectados e interaccionan con un gran número de nodos de menor jerarquía. La mayoría de los nodos tienen pocas uniones, pero existen hubs que se encuentran altamente conectados. Los hubs son puntos críticos que, en caso de eliminarse, determinan una gran disminución del tamaño de la red o su desintegración en subconjuntos (clusters o módulos) independientes (ver más adelante). La existencia de esos puntos críticos explica que las redes libres de escala sean altamente sensibles a modificaciones específicas (dirigidas a esos puntos críticos) pero muy resistentes a las distorsiones generadas al azar (como sucede generalmente en las mutaciones). Esta característica podría justificar por qué la susceptibilidad de una mínima proporción de las enfermedades complejas del ser humano (por ejemplo, diabetes, hipertensión arterial, neumonía) se asocia a mutaciones puntuales capaces de ser inducidas experimentalmente pero que muy difícilmente puedan ser inducidas mediante mutaciones generadas al azar. La gran mayoría de los sistemas biológicos corresponden a redes libres de escala.
Las principales redes libres de escala que existen en el ser humano son3:
- a)
Redes de unión para los factores de transcripción (transciption factor-binding networks). La utilización de microarrays y/o secuenciación de ADN en levaduras y otros organismos ha permitido identificar los sitios de unión de los factores de transcripción4,5. Posteriormente, y combinándolos con datos de expresión génica, fue posible deducir o identificar los sitios de activación y represión génica.
- b)
Redes de interacción entre proteínas (protein-protein interaction networks). Estas redes son las más estudiadas puesto que existen técnicas para identificar de forma masiva la presencia de proteínas (por ejemplo, mediante la combinación de electroforesis en gel de poliacrilamida bidimensional y la espectrometría de masas) y sus interacciones (por ejemplo, mediante la técnica de hibridación de 2 levaduras [«Y2H»]).
- c)
Redes de fosforilación (protein phosphorylation networks). La fosforilación de proteínas es un importante mecanismo regulador en las células eucariotas, estimándose que el 2% de los genes codifican enzimas con actividad cinasa6. Los avances en técnicas como la espectrometría de masas, la utilización de cinasas que solo actúan sobre nucleótidos marcados y el desarrollo de micromatrices han permitido el análisis masivo del fenómeno de fosforilación celular y la construcción de este tipo de redes.
- d)
Redes metabólicas (metabolic interaction networks). Clásicamente, el metabolismo se ha descrito como un conjunto de rutas metabólicas que, partiendo de un determinado sustrato inicial que sufre una serie de transformaciones enzimáticas, alcanza un producto final. Una red metabólica, por su parte, se refiere al análisis global de todo el metabolismo mediante la aplicación de ecuaciones diferenciales no lineales de catálisis e inhibición mutua. Una característica muy importante surgida de la comparación entre las redes metabólicas de distintos organismos fue que todas son muy similares, lo cual refleja las bondades del modelo adoptado (ver más adelante)7.
- e)
Redes de interacción genética (genetic molecule interaction networks). La combinación de 2 mutaciones (pero no cada una de ellas independientemente) en un individuo puede determinar modificaciones graves8. En los seres vivos la mayoría de las mutaciones no originan efectos letales si se producen individualmente puesto que (i) son generadas al azar y afectan principalmente a procesos de adaptación al medio y no a la supervivencia del individuo, y (ii) existe una gran redundancia en la función. Sin embargo, cuando se producen 2 mutaciones en una misma vía, el efecto puede ser mayor (por ejemplo, muerte del individuo o potenciación del fenotipo).
Los sistemas biológicos deben ser estables, es decir, mantener sus características fenotípicas a pesar de las distintas alteraciones (ambientales o genéticas) que actúen sobre ellos. Esta estabilidad, también denominada robustez, es una propiedad inherente a todos los sistemas biológicos y, como se ha demostrado claramente, ha sido favorecida por la evolución de las especies9.
La robustez de un sistema biológico es relativa, puesto que ningún sistema puede soportar cualquier tipo de alteración, y alguna puntualmente dirigida es capaz de desestabilizarlo. Por ello, la robustez de un sistema biológico está basada en una serie de características:
- a)
Redundancia. Se refiere a la presencia de vías alternativas capaces de sustituir una vía que falle, sin que el sistema se vea afectado. El ejemplo más habitual podría estar constituido por las citocinas, proteínas secretadas principalmente por las células del sistema inmune innato y adaptativo, encargadas de mediar múltiples funciones. Estas moléculas se destacan por su acción pleiotrópica (capacidad de actuar sobre múltiples tipos celulares) y redundante (muchas citocinas tienen el mismo efecto). Estas 2 cualidades brindan al sistema biológico una enorme robustez y podrían explicar, por ejemplo, el fracaso de todos los tratamientos inmunomoduladores aplicados a la sepsis hasta el momento actual10.
- b)
Control por retroalimentación. Se refiere al control ejercido por «circuitos» mediante los cuales los productos permiten controlar el procesamiento de los sustratos. Es una forma efectiva de mantener la estabilidad de un sistema. La retroalimentación negativa o control termodinámico (el aumento de los productos inhibe el procesamiento de los sustratos) se utiliza principalmente para mantener el estado estable del sistema, mientras que la retroalimentación positiva o autocatálisis (el aumento de los productos estimula el procesamiento del sustrato) se utiliza para los procesos de sensibilización.
- c)
Modularidad. Los sistemas biológicos están conformados por unidades funcionales o módulos que son entidades semiautónomas que muestran un alto coeficiente de agrupamiento interno (fig. 1) pero con pocas conexiones con el resto de los nodos. La modularidad permite «encapsular» funciones, lo cual es muy beneficioso para la robustez del sistema así como para la evolución de las especies. Actualmente está demostrada la existencia de módulos metabólicos11, de interacciones proteína-proteína12 y de regulación génica13. Es fundamental tener presente que algunos módulos pueden activarse o inhibirse en determinadas circunstancias, por ejemplo ante estímulos externos como son las infecciones14.
- d)
Protocolización y jerarquización. Estas características son fundamentales para proporcionar orden, organización y coordinación al sistema. La protocolización implica tener un grupo de reglas que permiten una organización y coordinación eficientes de las distintas partes dentro de un sistema. La jerarquización, que es una parte dentro de la protocolización, se refiere a la asignación de distintos niveles de organización con el propósito de reducir los costes de la transmisión de información15. Como se ha mencionado más arriba, las redes biológicas son en su mayoría del tipo libre de escala, con la particularidad de que también son modulares y jerarquizadas (fig. 1).
Las características de las redes biológicas colaboran en la explicación del concepto, propuesto por nuestro grupo, de paradoja terapéutica, que se refiere al frecuente hecho de que intervenciones terapéuticas, eficaces en estudios en modelos animales, son ineficaces en ensayos clínicos. El estudio de nuevos tratamientos de numerosas enfermedades complejas (por ejemplo, el SDRA, la sepsis grave, o el shock séptico) genera con frecuencia resultados positivos en condiciones experimentales. Sin embargo, muy pocos de esos avances se confirman en ensayos clínicos. Una explicación posible de este hecho podría ser que los modelos se desarrollan en animales sanos (sin comorbilidad), genéticamente similares, jóvenes y sometidos a un daño específico. Este es el caso, por ejemplo, de modelos de lesión pulmonar aguda inducida mediante la administración intratraqueal de lipopolisacárido o mediante ventilación mecánica utilizando un volumen corriente elevado, o de modelos de sepsis inducida mediante ligadura y punción del ciego. En estos modelos, los mecanismos de respuesta del huésped se concentran en un grupo relativamente limitado de vías (módulos redundantes y jerarquizados), por lo que se maximiza la potencial eficacia del tratamiento. Otra diferencia esencial de los estudios en animales y en humanos es la gran similitud genética que existe entre los animales de laboratorio en comparación con la que existe en pacientes. Los animales provienen en general de la misma familia y se originan de cruces endogámicos, mientras que los humanos descienden de poblaciones relativamente panmícticas. No existe la panmixia absoluta (elección de la pareja reproductiva independiente del fenotipo), pues los rasgos fenotípicos tienden a agruparse.
Sin embargo, en la vida real el SDRA o la sepsis grave son el resultado de un amplio abanico de daños, posiblemente reiterados en el tiempo (por ejemplo, una peritonitis que requiere repetidas laparotomías), sobre todo en individuos de edad avanzada o inmunosuprimidos, con una gran heterogeneidad genómica y epigenómica (la epigenética se refiere a las modificaciones reversibles a nivel de la secuencia de ADN que están determinadas por la interacción huésped-ambiente). Estas características determinan que la respuesta del sistema biológico «huésped» sea muy diversa y heterogénea, participando gran cantidad de hubs y módulos que ponen en cuestión el efecto beneficioso observado en los modelos animales. Por ejemplo, fármacos inmunomoduladores como los anticuerpos anti-TNFα16, o los bloqueadores de la óxido nítrico sintasa17, han demostrado efectos beneficiosos en modelos animales, pero no cambian o aumentan la mortalidad en humanos, lo que podría deberse a que el bloqueo específico de un determinado módulo (vía) genera un mayor desequilibrio en la respuesta global del sistema.
Conservación de las redes biológicasOtra de las principales características de las redes biológicas es su alta conservación entre las distintas especies. Jeong et al.7 compararon bioinformáticamente la redes metabólicas de 43 organismos distintos y pertenecientes a los 3 dominios de la vida (eucariotas, bacterias y arqueas) obtenidas desde el repositorio WIT18. El mencionado repositorio predice la existencia de vías metabólicas en función de la anotación de los genes y de datos experimentales. Este estudio permitió alcanzar importantes conclusiones que se resumen a continuación:
- (i)
Confirmó que la gran mayoría de las redes metabólicas son de tipo sin escala.
- (ii)
La distancia media en las 43 redes fue de 3,2 lo cual significa que, en promedio, se necesitarían 3 pasos para convertir un metabolito en cualquier otro. Evidentemente, esta conclusión no es acorde a los datos experimentales, por lo que posteriormente Ma y Zeng19 reconstruyeron las redes metabólicas en 80 organismos distintos pero, a diferencia de lo realizado por Jeong et al.7, no incluyeron los metabolitos muy frecuentes ni las moléculas muy pequeñas como el ATP y el NADH. Encontraron entonces que la distancia promedio en las redes metabólicas de células eucariotas, bacterias y arqueas es de 9,57; 8,50 y 7,22, respectivamente, lo cual es más similar a las distancias obtenidas experimentalmente.
- (iii)
El diámetro de las redes metabólicas aumenta conforme se incrementa la complejidad del organismo en estudio. Sin embargo, este aumento no se produce de una forma exponencial, como sería esperable si las conexiones fuesen al azar. Este hallazgo apoya la hipótesis de que las conexiones se realizan siguiendo la teoría de las potencias (ver más adelante). Otra importante consecuencia de este tipo de crecimiento dirigido es que al aumentar la conectividad de algunos nodos, aumenta su importancia, y su eliminación conduce a la desintegración de la red en «clusters» disfuncionales. Se ha demostrado que la lista de los nodos más conectados en los 43 organismos analizados es prácticamente la misma. Estos nodos se refieren a un grupo muy reducido de metabolitos utilizados para procesos fundamentales y comunes a todos ellos. El resto de los metabolitos, es decir, los menos conectados, son los utilizados para adquirir las características distintivas de cada especie. Ma y Zeng19, complementando el trabajo de Jeong et al.7, postularon la estructura teórica macroscópica de las redes metabólicas con forma de «pajarita» (bow-tie) (fig. 1).
Se han propuesto varios modelos para explicar el crecimiento de las redes biológicas. El más aceptado es el de nodos con un grado fijo que son constantemente agregados y conectados a la red. En este modelo la probabilidad a priori de establecer una nueva conexión con un nodo determinado se incrementa conforme aumenta el número de conexiones que ese nodo tenía previamente20,21. Es el modelo de vinculación preferencial (preferential attachment model) de Matthew6,22. Por ejemplo, si existiesen 3 nodos denominados a, b y c, con 3, 10 y 15 conexiones, respectivamente, lo más probable es que el nodo c reciba la nueva conexión dado que es el más grande.
Se ha sugerido que (i) los nuevos nodos en las redes biológicas aparecen por duplicación génica, puesto que genes ortólogos (genes evolutivamente antiguos que están presentes en distintos organismos pero que codifican proteínas con la misma función) tienden a generar nodos con un mayor número de conexiones23; y (ii) la especialización de los nodos es relativamente rápida, pues las conexiones existentes entre genes parálogos (genes que se originaron por duplicación en la misma especie pero evolucionaron independientemente, por lo que codifican proteínas con funciones similares pero no idénticas) son distintas. Por esto se ha propuesto una segunda teoría denominada de dinámicas de unión (link dynamics), según la cual la evolución de una red se basa, además de en conexiones preferenciales, en la ganancia y pérdida de conexiones entre nodos específicos.
Estas teorías de crecimiento de las redes biológicas explican el funcionamiento de otros tipos de redes. Por ejemplo, en los negocios es más probable establecer un nuevo negocio si se genera una «conexión» con el gerente de la empresa que con el dependiente, aun cuando es posible que luego se generen otras conexiones y se pierda o reduzca la primera del gerente. Otro ejemplo es la vida social. Es más probable integrarse a un nuevo grupo de personas si se establece la conexión con el chico o chica más popular, aun cuando es posible que las conexiones posteriores, una vez incluido en la red, se estrechen con un individuo no tan popular pero que genera un interés en particular. Estos enlaces a su vez no son fijos y están influidos por factores ambientales. Basta observar las relaciones existentes entre los integrantes de cualquier laboratorio de investigación o de cualquier servicio hospitalario.
Genome-scale metabolic modelsAnalizar desde un punto de vista global los patrones obtenidos experimentalmente provenientes de las ciencias «-ómicas» requiere la construcción de modelos matemáticos denominados genome-scale metabolic models (GEM) en los cuales se integran los datos experimentales (patrón metabolómico) con la predicción de las interacciones que entre ellos ocurren mediante la utilización de plataformas bioinformáticas y de información procedente de grandes repositorios y bases de datos24-26.
Existen varios tipos de modelos metabólicos o GEM, aunque todos tienen en común el objetivo de identificar matemáticamente los puntos de restricción. Estos puntos de restricción permiten distinguir entre lo posible y lo imposible desde el punto de vista metabólico. Las restricciones son de 3 tipos:
- (a)
fisicoquímicas, definidas por (i) la ley de conservación de la masa y de la energía, (ii) la dependencia entre las reacciones y la concentración de los metabolitos, y (iii) el cambio en la energía libre de las reacciones espontáneas;
- (b)
ambientales, por ejemplo, la disponibilidad de nutrientes; y
- (c)
regulatorias, por ejemplo, cómo la célula se adapta a los cambios ambientales.
La construcción de un GEM comienza con la elaboración de una matriz estequiométrica. Describir exhaustivamente el proceso de generar una matriz estequiométrica excede el propósito de la presente revisión (ver Covert et al27). Simplemente mencionaremos que el proceso comienza con la recolección de datos de la literatura (por ejemplo, desde Uniprot [www.uniprot.org], BRENDA [www.brenda-enzymes.info], Biocyc [www.biocyc.org] o KEGG [http://www.genome.jp/kegg]) para generar un listado de productos químicos y reacciones de transporte junto con los metabolitos que participan en ellas en una determinada célula. En una matriz estequiométrica cada columna corresponde a un elemento químico o reacción de transporte, con valores distintos de cero para identificar qué metabolitos participan en la reacción, así como el coeficiente estequiométrico que corresponde a cada metabolito. La matriz debe tener direccionalidad (los substratos tienen coeficientes negativos y los productos coeficientes positivos). Las filas de la matriz conforman la lista de reacciones en la cual un metabolito participa.
El balance de masas (mass balance) constituye una aplicación de la ley de conservación de masas en un sistema fisicoquímico con el objetivo de identificar los flujos o reacciones que suceden mediante el análisis de lo que ingresa y sale en un determinado sistema. Actualmente las matrices estequiométricas también pueden unirse con datos «-ómicos», provenientes, por ejemplo, del estudio del transcriptoma. Se puede conectar, aunque existen gran cantidad de dificultades, cada elemento químico o reacción de transporte a las proteínas o genes que permiten la formación del elemento o la reacción, con el propósito de mejorar la representación del sistema.
Metabolómica y genome-scale metabolic modelsLa metabolómica se refiere al estudio global de los metabolitos endógenos, y representa una «foto instantánea» de la expresión génica y de la actividad enzimática y fisiológica22,28–30 de un sector específico (por ejemplo, un tejido o la sangre) de un individuo. La metabolómica se origina a partir del concepto de que las enfermedades humanas están asociadas a un estado metabólico anormal y, por tanto, se reflejan en cambios a nivel de los componentes de los tejidos y líquidos corporales31,32.
La identificación masiva de los metabolitos puede ser realizada en células, tejidos o fluidos biológicos, utilizándose principalmente 2 técnicas: (a) espectroscopia de alta resolución mediante resonancia magnética nuclear; y (b) espectrometría de masas en tándem con electroforesis capilar, cromatografía de gases, cromatografía líquida, y espectrometría de masas transformada de Fourier de resonancia iónica en ciclotrón. Ambas modalidades se consideran complementarias.
El análisis metabolómico permite (a) identificar metabolitos; (b) cuantificar su concentración; (c) relacionar los metabolitos con situaciones especiales, definiendo así biomarcadores con un valor diagnóstico, pronóstico o de respuesta al tratamiento; (d) comprender la función de los metabolitos en vías metabólicas particulares o en toda la red metabólica en general, proporcionando así información sobre la patogénesis de una entidad; (e) identificar dianas terapéuticas; y (f) obtener imágenes de forma no invasiva, como la imagen de resonancia magnética nuclear o la tomografía de emisión de positrones o PET (de su acrónimo en inglés).
El primer ser vivo en ser modelado fue el Haemophilus influenzae33. Actualmente, existen numerosos procariotas y eucariotas cuyas redes biológicas han sido reconstruidas mediante esta aproximación holística34,35. En humanos, en el momento actual, se han publicado 3 GEM denominados Recon 136, Red metabólica humana de Edimburgo37 y HumanCyc38.
El Recon 1, al cual nos referiremos de forma particular por ser el más ampliamente utilizado, fue publicado en 200736 e incluye 1.496 marcos abiertos de lectura (en ingles open reading frames; se refiere a secuencias de ADN que codifican para proteínas), 2.004 proteínas, 2.712 metabolitos y 3.311 reacciones metabólicas.
Actualmente, las principales aplicaciones del Recon 1 son:
- (a)
Integrar las ciencias «-ómicas» para la construcción de modelos específicos. Por ejemplo, se pueden integrar estudios de transcriptómica o proteómica mediante el Recon 1 para determinar las reacciones en una célula o tejido en particular.
- (b)
Mapear genes homólogos para la construcción de modelos. La utilización de animales de experimentación es esencial en el desarrollo científico, por lo cual se han desarrollado modelos restrictivos para animales39,40. Sin embargo, resultan muy poco exactos. Recientemente, se han utilizado algoritmos para la adaptación del Recon 1 a modelos animales aprovechando la gran homología de secuencia que existe entre determinados animales41,42.
- (c)
Interpretar los datos obtenidos de forma masiva en los estados de enfermedad o en la respuesta a los tratamientos.
- (d)
Simular estados patológicos o respuesta a tratamientos.
Recientemente Agren et al.43 han publicado un protocolo para la elaboración automática de los GEM en líneas celulares específicas. El algoritmo fue aplicado a 69 tipos celulares humanos distintos y 16 tipos de cánceres.
El análisis de agrupamiento («clusters») jerárquico no supervisado (sin conocer o definir previamente de qué grupo se trata) de las 69 redes celulares y las 16 de cáncer encontró que entre las células normales existe una tendencia a agruparse en función de la cercanía anatómica que presenten (por ejemplo, las células de la corteza y médula adrenal se juntan). De forma inesperada, se encontró que las células cancerígenas pueden diferenciarse en 3 grupos: (a) cáncer de hígado, colorrectal, mama y endometrio; (b) cáncer de cuello y cabeza; y (c) el resto. Adicionalmente, se encontró que solo 189 reacciones (el 4,1% del total de las reacciones) son exclusivas para una línea celular o cáncer específico, mientras que 501 (el 11% del total de las reacciones) son comunes a todas las células. También compararon las redes de los 16 tipos celulares de cánceres con los 24 tipos celulares normales de los cuales provienen y concluyeron que: (a) existe un aumento pronunciado del metabolismo de las poliaminas (espermidina, espermina y putrescina), cuyos efectos incluyen función antioxidante, supresión de la necrosis y aumento de la proliferación celular; (b) aumento en la vía de la síntesis del isoprotenoides en especial del difosfato de geranilgeranil, implicados en varios eventos prooncogénicos; y (c) aumento en la producción de prostaglandinas, leucotrienos y ácido hidroperoxieicosatetranoico, compuestos con importante participación en la inflamación y estimulación de la progresión del cáncer.
Una visión de futuroA pesar de los recientes avances, la capacidad de analizar datos de forma masiva e integrarlos mediante una visión holística aún es insuficiente. El futuro próximo experimentará un avance tecnológico significativo con un incremento en la sensibilidad y especificidad para detectar muchos más metabolitos y otras moléculas, así como un drástico descenso en los costes de análisis y procesamiento. Estos cambios permitirán el estudio de los sistemas biológicos en distintas condiciones de salud y enfermedad, así como en tiempos evolutivos diferentes (nacimiento, adolescencia y senectud, o al inicio y al final de la patología, por ejemplo).
Se deberán mejorar los sistemas de detección de interacciones de moléculas. Por ejemplo, se dispone de técnicas para analizar masivamente las interacciones entre proteínas (two-hybrid screening) pero no para metabolitos, lo cual constituye una de las grandes dificultades actualmente.
Asimismo, es necesario el desarrollo de plataformas bioinformáticas mejores y más potentes, capaces de incluir, además de los metabolitos y la expresión génica, otras potenciales «restricciones», por ejemplo, patrones epigenéticos y de microARN.
La posibilidad futura de disponer de datos de forma masiva plantea el desafío de la transformación de esa información en conocimiento útil. En este contexto, la interpretación intuitiva o basada en la experiencia del investigador pierde validez, haciéndose necesaria la utilización de herramientas como la minería de datos (data mining) que permitan la identificación de patrones o elementos relevantes en conjuntos grandes de datos. Se trata de la misma aproximación que la que es requerida para el análisis de textos (text mining) en grandes bases de datos (por ejemplo, Pubmed). Los algoritmos que la minería de datos en general o la minería de textos en particular utilizan son extremadamente complejos (por ejemplo, redes neuronales, árboles de decisión, agrupamiento o reglas de asociación). Se han desarrollado programas de uso libre (por ejemplo, Weka, http://www.cs.waikato.ac.nz/ml/weka/) que son sencillos de utilizar y cuyos resultados son altamente fiables. A pesar de lo mencionado, siempre resulta necesaria una validación experimental final de los resultados obtenidos mediante estas estrategias.
Finalmente, un aspecto básico para la correcta interpretación de los resultados es el carácter multidisciplinario de los equipos de investigación. La colaboración entre expertos en bioinformática y expertos en el área concreta de estudio debe ser temprana en el planteamiento del diseño del estudio y debe mantenerse a lo largo del desarrollo de la investigación.
ConclusionesLa ciencia moderna ha presentado desde sus inicios una constante transformación impulsada en gran medida por el desarrollo tecnológico. El siglo pasado fue el auge del enfoque reduccionista, en el cual se plantea describir las «partes» de cada sistema y −en función de ellas− deducir el funcionamiento general. En el presente siglo, la irrupción de tecnologías de alto rendimiento asociadas al desarrollo de las plataformas bioinformáticas permitió el análisis de los procesos biológicos como un «todo» y no como la suma de sus «partes». Este nuevo enfoque se ha denominado «holístico» o «biología de sistemas» y presenta características que le son propias y compartidas por la mayoría de los organismos. En la biología de sistemas lo importantes es cómo todos los elementos interactúan entre sí, y se relega la importancia de las interacciones específicas entre uno o pocos elementos. La aplicación de la biología de sistemas a procesos o problemas vinculados a la salud ha originado una nueva disciplina denominada medicina de sistemas.
Los diagramas de grafos son una manera de representar las interacciones entre los elementos pertenecientes a una red biológica. Casi todas las redes pertenecientes a los seres vivos son del tipo «libre de escala».
Actualmente resulta posible la integración de los datos producidos de forma masiva mediante modelos matemáticos complejos basados en la realización de matrices estequiométricas y en la identificación de los puntos de restricción. Sin ignorar los desafíos y las dificultades que se presentarán en el futuro, la perspectiva holística plantea una oportunidad excepcional para la comprensión de los procesos biológicos de una forma integrada y para el desarrollo de tratamientos innovadores.
FinanciaciónSéptimo Programa Marco de la Unión Europea (ITN-FP7-26486).
Instituto de Salud Carlos III (FIS PI 12/2898)
Instituto de Salud Carlos III (FIS PI 11/2791)
PC es becario del Programa Marie Curie (PI-NET: ITTN 264864).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.







