



A recent systematic review and meta-analysis has reported the association between long-term use of angiotensin-converting enzyme inhibitors (ACEIs) and the risk of lung cancer (LC). Of the 2400 records reviewed, 13,061,226 patients met the inclusion criteria.1 The aim of this correspondence is to explain the underlying mechanism that could be involved in the increased risk of LC mediated by ACEIs. Substance P peptide (SP), mediator of neurogenic inflammation and neurokinin-1 receptor (NK-1R) could be considered mechanistically responsible for the induction of carcinogenesis and LC mediated by ACEIs for the following reasons: ACEIs hydrolyze SP,2 while ACEIs can prevent the degradation of SP by blocking ACE.2 ACEIs increase SP levels and induce cough,3 in fact, ACEIs to cause coughing in 5–10% of patients,3 also induce neurogenic inflammation4 and LC progression4,5 (Fig. 1). ACE or kininase II is a peptidyl dipeptidase which cleaves C-terminal dipeptides from a wide variety of substrates, such as angiotensin I, bradykinin, Met-enkephalin, neurotensin and SP.2 It is well known that ACE converts the inactive decapeptide angiotensin I to the octapeptide angiotensin II. However, an important and less known fact is that ACE also hydrolyzes SP, leading to an increase in SP levels.2,3 In contrast, the hydrolysis of SP was completely blocked by ACEI captopril.2 Thus, ACEIs therapy increases SP levels and induces cough3 (Fig. 1). SP plasma levels are higher in subjects with cough3 (including patients with ACEIs treatment) and in patients with cancer compared to healthy subjects.5 The above studies point to the fact that the use of ACEIs for many years could lead to increased levels of SP in lung tissues, possibly resulting in ensuing neurogenic inflammation.4 Persistently elevated SP levels could be playing a role in cancer promotion through the induction of SP-mediated neurogenic inflammation, furthermore it is well known that chronic inflammation is the hallmark of cancer induction.4 Moreover, SP increases epithelial cells proliferation through NK-1R4 and increased mitogenesis, in turn, increases rates of mutagenesis and thus carcinogenesis, ultimately leading to LC. Furthermore, it has been reported that: LC cells overexpress SP and NK-1R, NK-1R is essential for LC cell viability, and that SP induces LC cell proliferation in a concentration-dependent manner.4 In contrast, specific NK-1R antagonists including aprepitant drug (used in chemotherapy induces nausea and vomiting) in a concentration-dependent manner have: (1) cough suppressant effects in LC patients,5 (2) could counteract SP-mediated promotion of carcinogenesis through anti-inflammatory effects,4 (3) inhibits LC cells proliferation, induces apoptotic in LC cells, inhibits angiogenesis and inhibits tumour cell migration (counteracting invasion/metastasis).4,5
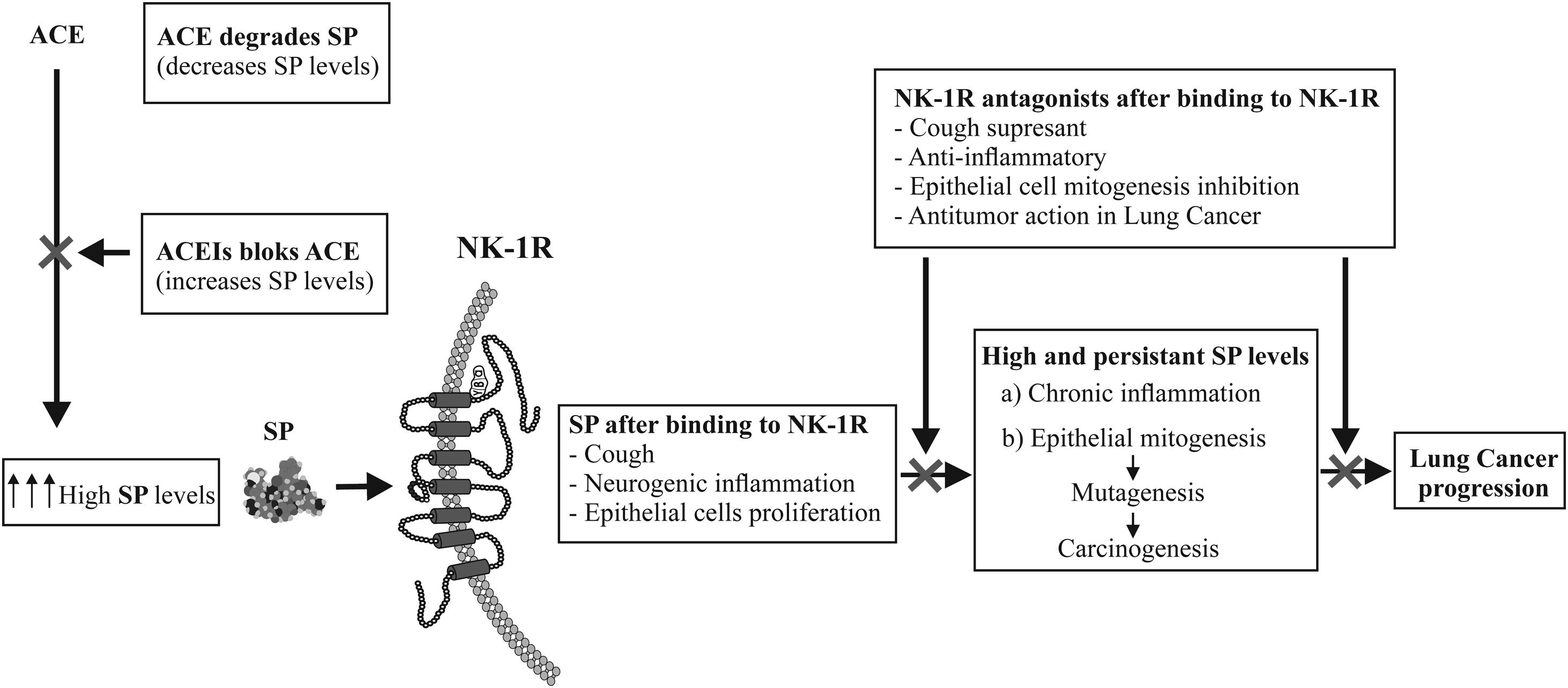
In summary, the use of ACEIs and its association with risk of LC is supported by recent epidemiological evidences. Furthermore, the rationale for etiopathogenesis of ACEIs in carcinogenesis of LC is supported by underlying mechanisms involving the SP/NK-1R system (ACEIs blocks ACE and increases SP levels, which after binding to NK-1R can induce cough and when used long term could cause LC). Thus, the long-term therapeutic use of ACEIs should be assessed by new studies to determine the scope of this rationale.
Authors’ ContributionsAll authors participated equally.
FundingNot declared.
Competing InterestsNot declared.
We would like to thank Mr. Javier Muñoz (Seville University) for technical assistance.







