

Over the past decade it has become increasingly evident that endogenous danger signals, called damage associated molecular patterns (DAMPs), released from damaged or dying lung resident cells play a pivotal role in the pathophysiology of chronic obstructive pulmonary disease (COPD).1,2 It has been consistently shown that the levels of DAMPs are increased in the lungs of COPD patients compared to non-COPD controls, in bronchoalveolar lavage fluid, epithelial lining fluid or sputum,3–5 as well as systemically in serum or plasma.6–8 These increased DAMP levels originate from structural and immune cells that are exposed to damaging agents upon inhalation of toxic gases and particles, like cigarette smoke, exhaust fumes or air pollution.9,10 Previously, we showed that the amount and type of DAMPs that are released upon inhalation of cigarette smoke, has a strong genetic component.11–13 However, to date it is still unknown which genetic factors increase the susceptibility for DAMP release. This study aimed to increase our understanding of the factors involved in susceptibility to DAMP release, which are potentially contributing to the susceptibility to develop COPD. This is the first study assessing the association between genetic factors and the levels of DAMPs in serum.
To this end, data was collected from 165 severe COPD (GOLD stage 3–4) patients of the Groningen Severe COPD cohort (NCT04023409), who were all ex-smokers with >5 pack-years of smoking history but have not smoked for >6 months prior to sampling. Forty-nine of the subjects were male with a mean age of 55 years (between 36 and 77), an average number of pack-years of 36.7 (SD=15.0), average lung function of 28 (SD=6.7; FEV1/FVC%), and a mean emphysema score of 40.2% (SD=7.5; %voxels <950 Hounsfield Units upon expiration). The study was approved by the medical ethical committee of the University Medical Center Groningen, and all participants provided written informed consent.
A panel of 4 DAMPs (α-Defensin, Galectin-9, sRAGE, dsDNA) was measured in serum of 165 COPD patients. The levels of α-Defensin (Human alpha-Defensin 1 DuoSet ELISA, DY8198-05, R&D Systems, Minneapolis, MN), Galectin-9 (Human Galectin-9 DuoSet ELISA, DY2045, R&D Systems) and the soluble Receptor for Advanced Glycation End-products (sRAGE; Human RAGE DuoSet ELISA, DY1145, R&D Systems) were measured by ELISA according to the manufacturer's protocols. The levels of double-stranded (ds)DNA were measured using Quant-iT™ PicoGreen™ dsDNA Assay Kits (P7589, Invitrogen, Waltham, MS). Whole-exome sequencing was performed on blood DNA using the Illumina platform and subsequently mapping the sequence to the Human Genome build 38. Full-genome gene expression data was obtained from bronchial brushes from 123 of the donors. The number of rare variants within a gene was assessed with the burden test using the sequence kernel association test (SKAT) method.14p-Values were computed using the SKAT_Null_Model function, in which each DAMP was defined as a continuous trait, and correction for age and smoking history was applied. Multiple testing correction was performed using the Bonferroni correction.
To test whether the levels of DAMPs in serum associated with the amount of potentially damaging rare variants of a gene, a genome-wide burden test was performed. This test assessed the associations between all variants within a gene and the serum levels of DAMPs. Manhattan plots indicate the different genes and their genomic location that were found to be significantly associated with the levels of dsDNA, Galectin-9, sRAGE and α-Defensin in serum (Fig. 1A–D). Nine genes were identified containing significantly more rare variants in COPD patients with high serum levels of dsDNA (HHLA2, ORM1, MROH6, SYT7, GJB4, HEATR6, OR2V2, TF, PLEK2). Eleven genes contained significantly more rare variants in COPD patients with high serum levels of Galectin-9 (TRAPPC2L, LYZ, C8B, RGS9, SOAT2, ZBTB32, PDE11A, OR5H6, HAPLN3, IGLV5-45, ANKRD22), while two genes contained more rare variants in COPD patients with high levels of sRAGE (FAM175A, AARS2). No genes were associated with the serum levels of α-Defensin. No obvious molecular pathways or biological functions were found to be related to the identified genes upon performing gene ontology enrichment analysis (The STRING database, http://string-db.org).
 dsDNA, (B) Galectin-9, (C) sRAGE, and (D) α-Defensin and the number of variants within a specific gene. The genomic location, and corresponding chromosomes are shown on the x-axis and the log transformed p-values are shown on the y-axis, a corrected p-value higher than −log10(p) was considered statistically significant (presented as a horizontal line). (E) Association between the serum levels of Galectin-9 and broncho-epithelial gene expression of KIF18A. Pearson correlation coefficient (r) and associated p-value are shown.")
Association between serum DAMP levels and the number of rare variants within a gene. A genome-wide burden test was performed to test the association between the serum levels of (A) dsDNA, (B) Galectin-9, (C) sRAGE, and (D) α-Defensin and the number of variants within a specific gene. The genomic location, and corresponding chromosomes are shown on the x-axis and the log transformed p-values are shown on the y-axis, a corrected p-value higher than −log10(p) was considered statistically significant (presented as a horizontal line). (E) Association between the serum levels of Galectin-9 and broncho-epithelial gene expression of KIF18A. Pearson correlation coefficient (r) and associated p-value are shown.
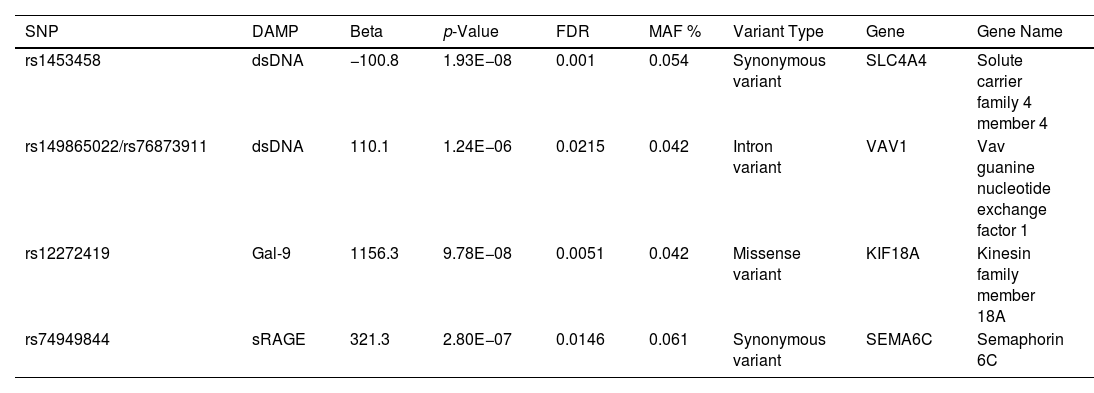
Next, the associations between serum levels of DAMPs and single nucleotide polymorphisms (SNPs) were assessed, using a protein quantitative trail locus (pQTL) analysis. Rare variants with a minor allele frequency of <4% were excluded. Table 1 shows the five SNPs that were identified to be significantly associated with the serum levels of DAMPs. Two of the SNPs were in full linkage disequilibrium (LD). The SNPs were located within the coding region of four different genes. No overlap between the identified pQTLs and genes containing more rare variants was found.
Significant Associations Between the Serum Levels of DAMPs and SNPs.
| SNP | DAMP | Beta | p-Value | FDR | MAF % | Variant Type | Gene | Gene Name |
|---|---|---|---|---|---|---|---|---|
| rs1453458 | dsDNA | −100.8 | 1.93E−08 | 0.001 | 0.054 | Synonymous variant | SLC4A4 | Solute carrier family 4 member 4 |
| rs149865022/rs76873911 | dsDNA | 110.1 | 1.24E−06 | 0.0215 | 0.042 | Intron variant | VAV1 | Vav guanine nucleotide exchange factor 1 |
| rs12272419 | Gal-9 | 1156.3 | 9.78E−08 | 0.0051 | 0.042 | Missense variant | KIF18A | Kinesin family member 18A |
| rs74949844 | sRAGE | 321.3 | 2.80E−07 | 0.0146 | 0.061 | Synonymous variant | SEMA6C | Semaphorin 6C |
p-Values represent the nominal p-value. A genome-wide false discovery rate (FDR) of <0.05 was considered genome-wide significant. Rs149865022 and rs76873911 are in full linkage disequilibrium.
Next, to test whether the identified SNPs affected the expression of the associated genes, broncho-epithelial gene expression of the four identified genes, i.e.SLC4A4, VAV1, KIF18A and SEMA6C, were correlated with the levels of the associated DAMPs in serum. No correlation was found between serum dsDNA levels and SLC4A4 and VAV1 or between serum sRAGE and SEMA6C. However, a small but borderline significant correlation was found between the serum levels of Galectin-9 and bronchial gene expression of KIF18A (Fig. 1E).
This is the first study investigating the genetic factors involved in susceptibility to DAMP release. We identified five SNPs, located within the coding region of four genes that are associated with the levels of DAMPs in serum of COPD patients. Out of these SNPs, only rs12272419 within the KIF18A gene is a missense variant leading to an amino acid change, potentially explaining why only the bronchial gene expression of KIF18A correlated with serum DAMP levels. KIF18A, a member of the kinesin superfamily, is involved in cell proliferation and mitosis, by regulating the dynamics of microtubule-associated molecular motors. Over-expression of KIF18A promotes cell proliferation and inhibits apoptosis.15 It is possible that in COPD, a disease where cell death processes like apoptosis, necroptosis and efferocytosis are dysregulated,16,17 further dysregulation of the cell death and proliferation mechanisms contribute to additional DAMP release. Likewise, sodium bicarbonate cotransporter 1 (SLC4A4), is also involved in the regulation of cell death, as it is involved in necroptosis, a regulated form of necrosis.18 Necroptosis has been shown to be one of the main contributors of DAMP release upon inhalation of cigarette smoke,19 and is increased in experimental and human COPD models.17
The amount of rare variants of specific genes was found to be associated with the levels of DAMPs in serum of COPD patients. No specific pathway or relationship between the identified genes was observed. Future studies should be focused on identifying the exact role of the increased number of rare variants within these genes and the levels of DAMPs in serum. It is likely that the increased number of mutations of these genes contribute to the release of DAMPs in the lungs upon cell damage, yet it is also possible that high levels of DAMPs in serum increases the mutation frequency of specific genes. The panel of DAMPs was selected to comprise a range of DAMPs originating from different subcellular compartments and that can activate different pattern recognition receptors. However, it is possible that a different selection of DAMPs would have led to the identification of different DAMPs.
Although we aimed to identify general susceptibility genes for DAMP release, we chose to use a cohort consisting exclusively out of COPD patients, since these patients have higher systemic levels of DAMPs. To this end, we cannot exclude the possibility that the identified susceptibility genes are related to DAMP release only in COPD patients. Future studies should be aimed at validating whether the DAMP release-related genes identified in this study are also involved in DAMP release in non-COPD patients.
In conclusion, this study confirmed that the susceptibility to the release of DAMPs has a strong genetic component. Several SNPs and susceptibility genes were identified that are associated with the release of DAMPs in COPD patients.
Ethical ApprovalThe study was approved by the medical ethical committee of the University Medical Center Groningen, and all subjects provided written informed consent. Severe COPD patients are derived from ClinicalTrials.gov Identifier: NCT04023409. Data from these study cohorts can be accessed through collaboration by contacting Maarten van den Berge (m.van.den.berge@umcg.nl).
FundingSimon D. Pouwels was funded by a Dutch Research Council (NWO) VENI grant (no. 09150162010003).
Authors’ ContributionsConcept & design: AF, SDP. Data analysis: FSB, AF, SDP. Patient inclusion/data collection: VB, IAE, MvdB, DJS, PMH. Project supervision: SDP. Manuscript preparation: SDP. Manuscript revision: FSB, VB, IAE, PMH, DJS, MvdB, AF, SDP.
Conflict of InterestNone declared.
Declaration of Artificial Intelligence InvolvementNo artificial intelligence was used for this study, nor for the writing of the manuscript.







