

Introducción
En el documento de consenso realizado recientemente por la American Thoracic Society (ATS) y la European Respiratory Society (ERS), la enfermedad pulmonar obstructiva crónica (EPOC) se define como "un proceso patológico que se puede prevenir y tratar, y se caracteriza por una obstrucción del flujo aéreo que no es completamente reversible. La limitación del flujo aéreo es, por lo general, progresiva y se asocia con una respuesta inflamatoria pulmonar anormal a partículas o a gases nocivos, principalmente causada por el tabaquismo. Aunque la EPOC afecta a los pulmones, también produce consecuencias sistémicas importantes"1. En la actualidad, es una de las principales causas de morbimortalidad en los países industrializados. Aunque el hábito tabáquico es su principal causa, sólo el 15% de los fumadores desarrolla la enfermedad, lo que indica que deben existir otros factores, tanto genéticos como ambientales, que interactúen entre sí, para dar lugar a una mayor susceptibilidad del sujeto a presentar la enfermedad. En una enfermedad compleja y multifactorial como la EPOC, en la actualidad se cree que, aunque determinadas alteraciones genéticas pueden condicionar la enfermedad, la asociación entre los polimorfismos génicos y el fenotipo probablemente no sea lineal, y el fenotipo final dependa de la genética, el ambiente y el entorno en el que ese genotipo se desarrolle2. Se han identificado varios factores de riesgo ambientales, como la contaminación medioambiental, las infecciones virales en la infancia y las infecciones latentes por adenovirus, así como varias alteraciones genéticas, que pueden aumentar el riesgo de presentar EPOC. En esta revisión se describen algunos de los principales factores implicados en la patogenia de la EPOC (tabla I).

Factores ambientales
Hábito tabáquico
El consumo de tabaco es el principal factor de riesgo para el desarrollo de EPOC. Los sujetos fumadores tienen mayor riesgo de descenso del volumen espiratorio forzado en el primer segundo (FEV1) que los no fumadores, con un rango de caída de este parámetro de entre 7 y 33 ml/año3. Este descenso está relacionado con la cantidad de tabaco consumido. Asimismo, se ha visto que el abandono del hábito tabáquico desacelera esta caída, aunque el FEV1 no recupera su valor basal.
Numerosos estudios han demostrado la relación entre el tabaco y la EPOC. Uno de los más importantes ha sido el realizado durante los últimos 50 años en el Reino Unido, en el que se ha estudiado a médicos de todo el país. Los resultados de los primeros 40 años de seguimientos mostraron que la mortalidad total era 2 veces mayor en los sujetos fumadores que en los no fumadores4. Este incremento de mortalidad era proporcional al consumo de cigarrillos. Al analizar la tasa de mortalidad global en sujetos de entre 35 y 69 años, ésta era del 20% en los no fumadores, y aumentaba al 41% entre los fumadores leves (1-14 cigarrillos/día) y al 50% en los grandes fumadores (≥ 25 cigarrillos/día). Resultados similares se obtuvieron a los 50 años de estudio: se constató que tanto el cáncer de pulmón como la EPOC tenían una relación estadísticamente significativa con el consumo continuado de tabaco y con el número de cigarrillos consumidos al día (p < 0,0001). La mortalidad también resultó mayor en el grupo de los fumadores; las causas más frecuentes fueron el cáncer de pulmón, la EPOC y la enfermedad isquémica cardíaca. Durante las últimas décadas del siglo xx, la longevidad en las personas no fumadoras ha aumentado considerablemente, debido a los avances de la medicina; sin embargo, esto no fue así entre los sujetos que continuaron fumando. Los fumadores morían una media de 10 años antes que los no fumadores5.
Exposición ambiental al humo del tabaco
La exposición al humo del tabaco desde edades muy tempranas aumenta el riesgo de presentar enfermedades respiratorias en la edad adulta, entre ellas la EPOC. El tabaquismo materno durante la gestación afecta al desarrollo pulmonar, como se ha visto en estudios realizados en animales, en los que la exposición de la madre al humo del tabaco influía negativamente en el desarrollo del pulmón fetal, y en estudios en neonatos de madres fumadoras en los que se ha observado una función pulmonar disminuida6. En la infancia, el hábito tabáquico en los padres, especialmente en la madre, aumenta la susceptibilidad de los niños a presentar infecciones del tracto respiratorio inferior.
El humo del tabaco inhalado de forma pasiva contiene muchas sustancias conocidas, probablemente carcinógenas, así como sustancias irritantes y tóxicas. Los resultados de un estudio reciente sobre la relación entre la exposición al humo del tabaco y la EPOC mostraron que los pacientes con un mayor tiempo de exposición tanto en el hogar como en el trabajo tenían un riesgo mayor de presentar EPOC, con independencia de otras variables7. Además, el exceso de riesgo relativo para los pacientes no fumadores con una exposición en el hogar elevada fue de 0,88, mientras que en los fumadores con escasa o nula exposición en casa fue del 1,83. Los pacientes fumadores con una elevada exposición dentro del hogar tenían un exceso de riesgo relativo de presentar EPOC de 4,32; es decir, mayor que la suma de ambos riesgos, lo que apoya la hipótesis de un efecto sinérgico de ambas exposiciones. El estudio prospectivo de Vineis et al8, realizado en no fumadores y exfumadores de más de 10 años, mostró un riesgo mayor, aunque no estadísticamente significativo, de presentar enfermedades respiratorias en estos últimos, respecto a los no fumadores.
Exposición laboral
En la clínica, no utilizamos el término EPOC ocupacional, como sucede con el asma, debido a que se trata de una enfermedad crónica, cuya obstrucción, irreversible, no se relaciona temporalmente con la exposición. Por tanto, la identificación epidemiológica de EPOC ocupacional se basa en constatar una prevalencia elevada de EPOC entre los trabajadores expuestos a determinadas sustancias9. En este sentido, existen numerosos estudios poblacionales que indican que una proporción de los casos de EPOC en la sociedad se deben a la exposición en el lugar de trabajo a polvos, humos, vapores y gases nocivos. Según la ATS, el riesgo atribuible poblacional (proporción de casos de una enfermedad debidos a una determinada exposición) de la EPOC debida a la exposición ocupacional se estima en alrededor del 15%. Los trabajadores de industrias textiles, plásticas, de gomas, madera, papel, así como los agricultores, mineros y trabajadores de la construcción tienen un riesgo aumentado de presentar EPOC por la exposición en el trabajo a sustancias, como grano, isocianatos, cadmio, carbón y otros polvos minerales, metales pesados y humos. La exposición ocupacional, además de aumentar el riesgo de EPOC, ha demostrado incrementar la mortalidad por esta enfermedad en los trabajadores expuestos, tanto fumadores como no fumadores6,9.
Además de estudios epidemiológicos, existen algunos estudios experimentales en animales que han demostrado que la exposición a determinados agentes produce enfisema. Algunos de estos agentes, como el cadmio, el carbón, la endotoxina y el sílice, ya se habían relacionado con el desarrollo de EPOC en estudios estadísticos previos9.
La prevención sigue siendo, hoy por hoy, nuestra mejor arma. Una adecuada historia clínica de nuestros pacientes nos orientará en la exposición laboral, si bien lo más importante es incidir en la necesidad de una adecuada prevención primaria en los centros de tra-bajo.
Contaminación ambiental
La contaminación ambiental se puede dividir en 2 grupos, según se localice en el exterior o el interior del hogar:
Exterior. La contaminación ambiental es un factor de riesgo para el desarrollo de EPOC. Varios estudios han mostrado la relación entre el grado de contaminación y la presencia de síntomas y enfermedad respiratoria. En el estudio SAPALDIA, los investigadores encontraron relación entre los valores de PM10 (partículas en suspensión con un diámetro menor de 10 mm) y NO2 exterior y una capacidad vital forzada (FVC) disminuida10. Además, se constató que la exposición de larga duración a la contaminación ambiental, incluso con bajos valores, se asociaba con una mayor prevalencia de síntomas respiratorios. Resultados similares se obtuvieron en Italia, donde se comprobó que los habitantes de zonas urbanas presentaban mayor riesgo de tener síntomas respiratorios crónicos que los de zonas rurales. Se han realizado otros estudios acerca de la exposición ambiental y la mortalidad en la población. Se estima que por cada 10 mg/m3 que aumenta la PM10, la mortalidad debida a enfermedades respiratorias y a enfermedades cardiovasculares aumenta un 3,4 y un 1,4%, respectivamente5. En Estados Unidos se han realizado varios estudios en los que se muestra un riesgo mayor de morir por enfermedades cardiorrespiratorias en las zonas con mayor contaminación medioambiental3.
Interior. Además del humo del tabaco, se han identificado otros posibles contaminantes que podrían producir daño en el aparato respiratorio. Estos contaminantes derivan de las calefacciones, las reacciones fotoquímicas, los muebles, los organismos biológicos y las fibras, y principalmente producen síntomas respiratorios, pérdida de función pulmonar, hiperreactividad bronquial e infecciones respiratorias6.
Estatus socioeconómico y estilo de vida
Diferentes estudios han demostrado la asociación entre el estatus socioeconómico y la EPOC. El nivel socioeconómico se valora por medio de indicadores como la renta anual, la educación, el tipo de trabajo, las características de la vivienda y el número de personas que habitan en la casa, respecto a su tamaño6. Los trabajos realizados en función pulmonar han demostrado una relación significativa entre un estatus socioeconómico bajo y una FVC y un FEV1 bajos, y se han encontrado diferencias en el FEV1 de 200 a 400 ml, entre los grupos más extremos. Si se tiene en cuenta el nivel educativo de los sujetos, aquellos con una educación primaria o secundaria tienen una prevalencia significativamente mayor de obstrucción en la espirometría que los que tienen estudios universitarios. Otros trabajos centrados en la sintomatología de la bronquitis crónica han obtenido resultados similares a los de la función pulmonar3.
Resulta difícil conocer la contribución de cada indicador de nivel socioeconómico al desarrollo de la EPOC. Diferentes trabajos han estudiado diferentes marcadores concretos. Así, en un estudio realizado en Brasil se encontró que una escolarización deficiente, junto con una casa pobre y una renta familiar escasa, se asociaban a la presencia de bronquitis crónica3. Otros estudios han demostrado la relación entre síntomas respiratorios y peak flow con unos índices de bajo estatus y con un ambiente empobrecido en el hogar a los 2 años de edad. En un estudio realizado en Dinamarca, tanto la educación como la renta anual se relacionaron con una función pulmonar disminuida, con independencia de la edad, lo que podría indicar que los procesos implicados están relacionados con sucesos acaecidos antes de que el individuo alcance su función pulmonar máxima al comienzo de la edad adulta3.
Los mecanismos por los cuales el estatus socioeconómico y el estilo de vida influyen en la función pulmonar se desconocen, pero se cree que el crecimiento pulmonar intrauterino, las distintas exposiciones a una edad temprana, las infecciones respiratorias en la infancia, el tabaquismo en la infancia y en la edad adulta, el tipo profesión, las características de la vivienda y la nutrición podrían estar implicados3.
Todos estos trabajos indican que las condiciones en las que se desarrolla el individuo desde la infancia más temprana influyen en su salud y en el riesgo de tener enfermedades respiratorias en el futuro, lo que convierte a la pobreza no sólo en un problema social, sino en un problema de salud pública que requiere consideraciones futuras por parte de los diferentes Gobiernos.
Dieta y consumo de alcohol
La desnutrición se asocia con frecuencia a la EPOC y puede exacerbar su sintomatología, al alterar la función de los músculos respiratorios, la tolerancia al ejercicio y la inmunocompetencia del sujeto. En los últimos años, ha aumentado la atención en el posible efecto beneficioso de algunos nutrientes en el desarrollo de la EPOC; entre ellos, las vitaminas C y E que, debido a su acción antioxidante, podrían contrarrestar el daño oxidativo producido por la exposición al humo del tabaco y a contaminantes ambientales. En este sentido, se ha visto que los sujetos con un consumo bajo de frutas frescas presentan un FEV1 unos 80 ml menor del esperado6. También se ha encontrado una asociación entre el aporte de magnesio y el FEV1, con independencia de la vitamina C. Asimismo, el consumo de ácidos grasos omega-3, inhibidores del metabolismo del ácido araquidónico, podría proteger del desarrollo de EPOC en fumadores. En un estudio reciente en adultos fumadores, se ha visto que los valores de FEV1 son mayores en las personas con un consumo mayor de pescado3. Por último, el abuso de alcohol se ha barajado como un posible factor de riesgo de EPOC.
Actividad física
El ejercicio físico regular modifica tanto el riesgo de presentar EPOC como su curso clínico. Hay estudios experimentales que indican un efecto antiinflamatorio derivado de una actividad física regular. En sujetos que realizan una media de 2,5 h semanales de ejercicio físico se ha comprobado un incremento de citocinas antiinflamatorias, como la interleucina (IL) 4, la IL-10 y el factor de de crecimiento de transformación β(TGF-β), así como un descenso de la proteína C reactiva (PCR) y de algunas citocinas proinflamatorias, como la IL-6 y el factor de necrosis tumoral α (TNF-α)11.
Recientemente, se han publicado los resultados de un estudio prospectivo aleatorizado de 10 años de duración sobre la relación entre el FEV1 y el ejercicio físico12. En el estudio se incluyó a 6.790 pacientes, con una edad media de 52 años. El 23% de ellos no eran fumadores, el 22% eran exfumadores y el 55%, fumadores activos. Se dividió a los sujetos en 3 grupos, según realizaran una actividad física baja (menos de 2 h/semana), moderada (2-4 h/semana) o alta (más de 4 h/semana o ejercicio más intenso). En general, el riesgo de presentar EPOC fue mayor en los fumadores activos que en los no fumadores o los exfumadores. La caída del FEV1 fue menor en el grupo que realizaba una actividad física moderada o alta, frente a los que realizaban un ejercicio de baja intensidad, pero sólo entre los sujetos fumadores. También fue menor el descenso de la FVC. Los sujetos con un grado moderado-alto de actividad física presentaban un riesgo reducido de presentar EPOC, frente al grupo de actividad física baja (odds ratio = 0,80, intervalo de confianza del 95%, 0,65-0,98; p = 0,03). Ajustando los resultados según la actividad física basal al inicio del estudio, los sujetos que redujeron su actividad durante el seguimiento experimentaron un incremento de la caída del FEV1 y un aumento del riesgo de EPOC, mientras que las personas que durante el estudio aumentaron su actividad física mostraron una reducción en el descenso de la función pulmonar y del riesgo de EPOC comparado con los sujetos que no cambiaron sus grados de actividad física.
Los mismos autores analizaron la mortalidad y los ingresos hospitalarios y obtuvieron resultados similares. Los sujetos que realizaban una actividad física baja, moderada o alta presentaron un riesgo menor de ingreso hospitalario y mortalidad por cualquier causa que aquellos que realizaban una actividad física muy baja13.
La importancia de estos resultados radica en el hecho de que, por el momento, no se puede actuar en la mayoría de los factores de riesgo de EPOC, pero sí se puede variar el grado de ejercicio físico y así mejorar tanto el riesgo como el pronóstico de la enfermedad.
Infecciones respiratorias en la infancia
En la infancia, el 20-30% de las infecciones respiratorias son virales, y de ellas el 50% se producen por el virus respiratorio sincitial (VRS). Numerosos estudios han demostrado la asociación entre las infecciones del tracto respiratorio inferior en los primeros años de vida y la presencia de EPOC o asma en la edad adulta6. En el asma, el proceso parece relacionado con la respuesta de las células T-helper CD4+ y con una producción elevada de IL-4, IL-5 y otras citocinas que podrían inducir un proceso inflamatorio con aumento de la producción de IgE. Sin embargo, todavía se desconoce el mecanismo por el cual las infecciones virales en la infancia aumentan la susceptibilidad a la EPOC14.
Infección latente por adenovirus
Las infecciones virales pueden persistir después de un cuadro de infección aguda por varios mecanismos. Durante la infección, el virus penetra en la célula y la utiliza para replicarse. El ADN viral puede integrarse en el ADN del huésped o bien formar un plásmido y, cuando el grado de replicación viral es bajo, la infección puede persistir más allá de la infección aguda15. Existen evidencias de que este tipo de infecciones latentes pueden influir en la respuesta inflamatoria celular. Algunos estudios han encontrado ADN de adenovirus y proteína adenoviral E1A en células epiteliales pulmonares de pacientes con EPOC mucho tiempo después de que los síntomas de la infección aguda hayan desaparecido. La proteína E1A actúa como un amplificador de numerosos genes del huésped y ataca sitios de unión de factores de transcripción. Estudios in vitro han constatado que células E1A-positivas producen un exceso de citocinas inflamatorias y de moléculas de adhesión de superficie (ICAM-1) relacionadas con el factor de transcripción NF-kß14. El hecho de que la EPOC tenga un componente inflamatorio sugiere la hipótesis de que una infección viral latente podría amplificar la respuesta inflamatoria pulmonar producida por el humo del tabaco.
Factores genéticos
Como ya se ha afirmado previamente, la EPOC es una enfermedad de etiología compleja en la que la interacción entre el genotipo y el entorno desempeña un papel muy importante. Los factores de riesgo endógenos que contribuyen al desarrollo de EPOC son diversos, entre ellos el sexo, la hiperreactividad bronquial y determinadas alteraciones genéticas. La patogenia de la EPOC es compleja y multifactorial, pero existen 3 teorías que podrían explicar los procesos que tienen lugar en esta enfermedad16:
Teoría proteasa-antiproteasa: indica un desequilibrio entre las proteasas que digieren la matriz extracelular y las proteínas antiproteasas que la protegen. Esta teoría se fundamenta en el hecho de que el déficit de alfa-1 antitripsina (AAT) se relaciona con una mayor frecuencia de EPOC de inicio temprano. También se han implicado otras proteínas, como las metaloproteinasas (MMP), la captesina B y las colagenasas.
Teoría oxidación-reducción: el desequilibrio entre oxidantes lesivos al tejido pulmonar y antioxidantes protectores podría producir un estrés oxidativo que conllevaría la activación de proteasas y la liberación de mediadores de la inflamación.
Inflamación: la tercera teoría relaciona las 2 teorías anteriores.
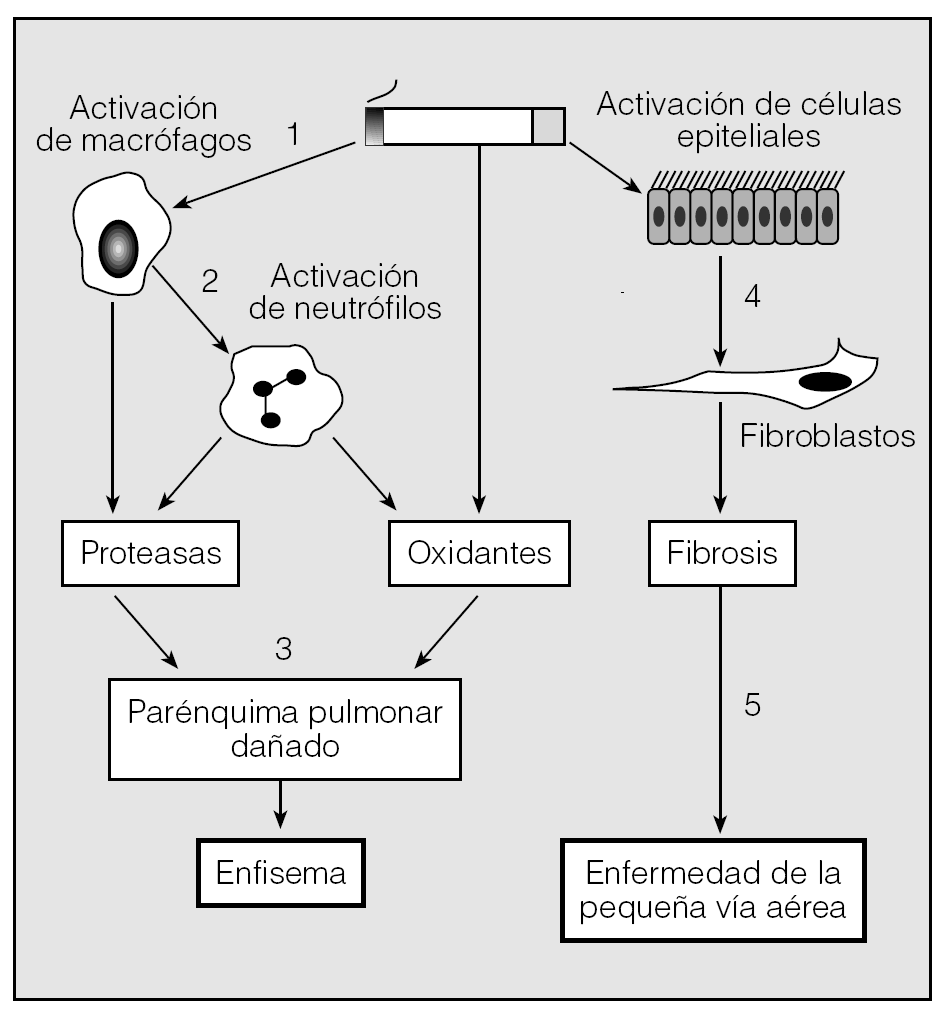
En la patogenia de la EPOC, el humo del tabaco desempeña un papel fundamental: por una parte, activa los macrófagos que liberan proteasas y activan los neutrófilos, y por otra, el tabaco activa las células epiteliales, promoviendo la actividad fibroblástica y produciendo fibrosis y enfermedad de la vía aérea pequeña. Los neutrófilos activados liberan tanto proteasas como oxidantes que dañan el parénquima pulmonar y dan lugar a enfisema (fig. 1).

Fig. 1. Fig. 1. La patogénesis de la EPOC. El humo del tabaco activa a los macrófagos (1) que liberan proteasas, sustancias quimiotácticas para los neutrófilos (2) y oxidantes que dañan el tejido conectivo pulmonar (3), causando enfisema. La estimulación de las células epiteliales activa a los fibroblastos produciendo lesión de la vía aérea pequeña (4 y 5). Modificado de Wood et al16.
Son muchas las proteínas implicadas en estos procesos. Cualquier alteración estructural en sus genes puede alterar estos equilibrios y producir enfermedad. Existen 2 cambios estructurales fundamentales: los microsatélites, que son repeticiones múltiples de un segmento corto de ADN, y los polimorfismos, que consisten en el cambio de una sola base del genoma. Si este cambio de base afecta a una región codificante y modifica la secuencia de aminoácidos de la proteína, probablemente se produzca enfermedad.
Sexo
Numerosos estudios han indicado que las mujeres tienen un riesgo mayor de presentar EPOC que los varones. Prescott et al17 observaron que la pérdida de función pulmonar asociada al tabaco era mayor en las mujeres. Si utilizaban los paquetes/año como indicador de la exposición al tabaco, el riesgo asociado al consumo era mayor en las mujeres que en los varones, aunque la diferencia no fue estadísticamente significativa. También se observó un riesgo mayor de ingreso hospitalario por la EPOC. El mecanismo por el cual esto sucede podría estar en relación con el hecho de que la hiperreactividad bronquial es más frecuente entre las mujeres3.
Hiperreactividad bronquial
En varios estudios se ha analizado el papel de la hiperreactividad bronquial en la patogenia de la EPOC. Existen 2 posibles teorías que implicarían la hiperreactividad bronquial. Por una parte, hay autores que sugieren que predispone a la pérdida de función pulmonar y a la EPOC, y otros, en cambio, mantienen que la hiperrespuesta bronquial podría ser el resultado de la inflamación de la vía aérea mediada por el humo del tabaco3. En varios trabajos se ha constatado que los pacientes con hiperreactividad bronquial presentan una función pulmonar disminuida. Se han descrito polimorfismos del receptor beta-2 implicados en la patogenia del asma grave, pero son necesarios más estudios para determinar su implicación en la EPOC18.
Alfa-1 antitripsina
La AAT es una glucoproteína que se engloba en el grupo de las antiproteasas. Es sintetizada mayoritariamente en el hígado y, en menor proporción, en los macrófagos alveolares y los monocitos de sangre periférica. Su principal función es inhibir la elastasa del neutrófilo de manera irreversible, protegiendo la matriz extracelular de su degradación. Además, puede inhibir la mayoría de las serinproteasas del neutrófilo, y neutraliza las defensinas alfa del neutrófilo, el leucotrieno B-4 (LTB-4) y la IL-8, mediadores que atraen al neutrófilo al foco inflamatorio19. El gen de AAT se localiza en el cromosoma 14 y se transmite por herencia mendeliana simple de manera autosómica codominante. Existen numerosos polimorfismos de este gen, la mayoría sin trascendencia clínica. Las 4 variantes genómicas más frecuentes, 3 de ellas debidas a mutaciones puntuales, se clasifican según su velocidad de migración electroforética: F (fast), M (medium), S (slow) y Z (very slow)16. El alelo M es el alelo salvaje y el más prevalente, presente en más del 90% de los sujetos sanos. El alelo Z se produce por la sustitución de una guanina por adenosina en el exón 5, cambiando glutamato por lisina en la proteína final. Es la deficiencia más frecuente en el norte de Europa, y en España este alelo está presente en el 1,5% de la población general20. En el alelo S se sustituye una adenina por timina en el exón 3, lo que conlleva el cambio de glutamato a valina en la proteína, y es la mutación más frecuente en el suroeste europeo. Estas 2 proteínas anormales son resistentes a la degradación enzimática, polimerizan en el hígado y quedan retenidas en el retículo endoplásmico de los hepatocitos el 80-90% de las moléculas AAT-Z y el 40-50% de las AAT-S21.
El déficit de AAT se clasifica por su genotipo y por su valor plasmático. Los valores séricos normales están comprendidos entre 120 y 220 mg/dl19. Las deficiencias de AAT pueden ser graves o intermedias, dependiendo de si el individuo es homo o heterocigoto para el gen alterado. El enfisema se produce porque las bajas concentraciones plasmáticas y tisulares de AAT no son suficientes para contrarrestar los daños producidos en el tejido pulmonar por las proteasas.
Los sujetos homocigotos ZZ tienen unos valores séricos de AAT aproximadamente del 10%22. Presentan un deterioro acelerado de la función pulmonar incluso en ausencia de tabaquismo. Hasta en el 60% de los casos pueden presentar obstrucción crónica del flujo aéreo, y el tabaquismo puede ser el principal factor de riesgo, ya que en fumadores el enfisema aparece a edades más tempranas. Sin embargo, este genotipo está presente sólo en el 1-2% de los casos de EPOC. El hecho de que no todos los individuos desarrollen enfisema indica que el déficit de AAT por sí solo no es suficiente para causar enfermedad y que deben estar presentes otros factores de riesgo, tanto genéticos como ambientales. Recientemente, se ha demostrado que determinados polimorfismos en el gen de la sintetasa del óxido nítrico endotelial (NOS3) y de la glutatión S-transferasa-P1 (GSTP1) podrían contribuir al desarrollo de enfisema en estos individuos18,19.
Las causas más frecuentes de déficit intermedio de ATT son los genotipos MS y MZ, presentes en el 10 y el 3% de la población caucásica, respectivamente. Los fenotipos MS y MZ presentan unas concentraciones séricas de AAT del 60 y el 40% de los valores normales, respectivamente. Se han realizado varios estudios sobre la prevalencia del fenotipo MS con resultados poco concluyentes, por lo que no se cree que suponga un riesgo incrementado de desarrollar EPOC22,19. En cuanto al fenotipo MZ, los resultados de estudios de casos y controles, y algunos en población general, indican que es un factor de riesgo para el desarrollo de enfermedad pulmonar, aunque el incremento del riesgo es pequeño y sólo en fumadores20. En el metaanálisis realizado por Hersh et al23, en los estudios de casos y controles, se observó un riesgo aumentado de EPOC para los sujetos MZ, aunque los datos no son concluyentes porque existía mucha variabilidad en la calidad y en el diseño de estos estudios.
Los individuos SS tienen unas concentraciones séricas de AAT del 60% con una prevalencia de EPOC similar a la población general. El fenotipo SZ es poco frecuente. Presenta unas concentraciones de AAT del 40% de los valores normales19. No se ha encontrado una mayor prevalencia de este fenotipo en pacientes con EPOC en estudios de casos y controles, aunque sí en estudios poblacionales, donde se demostró un aumento del riesgo de obstrucción crónica del flujo aéreo en estos sujetos, si eran fumadores18,20.
En el déficit de AAT existe gran variabilidad clínica incluso en sujetos con el mismo genotipo. El tabaquismo es un factor de riesgo para desarrollar enfisema en estos individuos, pero no explica toda la variabilidad existente. La exposición a irritantes profesionales, algunas ocupaciones de riesgo, las infecciones respiratorias y la presencia de sibilantes son otros factores que pueden condicionar la evolución de la enfermedad24. Por tanto, el diagnóstico temprano de estos pacientes puede ser fundamental a la hora de evitar los factores predisponentes que conllevan un mal pronóstico de la enfermedad.
Metaloproteinasas
Las MMP constituyen una familia de al menos 20 enzimas extracelulares con actividad proteolítica. Entre sus funciones se incluyen la degradación del colágeno, la inactivación de AAT y la activación del TNF-α16. Sus genes están localizados en los cromosomas 11, 14, 16, 20 y 22, y su sobreexpresión se ha relacionado con enfermedades como el enfisema y el cáncer.
Algunos estudios sugieren que varias de estas MMP, como la MMP1 (colagenasa-1), MMP12 (elastasa humana del macrófago) y la MMP9 (colagenasa B) podrían estar implicadas en la inflamación de la vía aérea y en la patogenia del enfisema18. En ese sentido, estudios experimentales en ratones han demostrado que la sobreexpresión de la MMP1 favorece el desarrollo de enfisema y que los ratones knockout para el gen de la MMP12 no desarrollan la enfermedad, incluso expuestos al humo del tabaco16,18. En pacientes fumadores con obstrucción crónica al flujo aéreo se han encontrado mayores concentraciones de MMP1 y MMP9 que en controles sanos, tanto fumadores como no fumadores16,21.
Se han descrito varios polimorfismos localizados en la región promotora de los genes de las MMP. El polimorfismo génico más estudiado es el de MMP9, en el cromosoma 20. La mutación en su promotor conlleva un aumento de su expresión, y hay estudios que han demostrado una asociación entre esta mutación y la EPOC16. La inserción de una guanina en la posición 1607 de la región del promotor de la MMP1 introduce un nuevo sitio de unión para el factor de transcripción, lo que se traduce en un aumento de la expresión de este gen. En el trabajo realizado por Joos et al25 en población caucásica fumadora, no se encontró asociación entre la pérdida de función pulmonar y ninguno de los genotipos evaluados (MMP1, MMP9, MMP12). Sin embargo, había una tendencia a la asociación (p = 0,05) entre los genotipos MMP1 y una función pulmonar disminuida. Los autores sugieren que los polimorfismos en los genes de MMP1 y MMP12, pero no en el de MMP9, son factores predisponentes del daño pulmonar causado por el tabaco.
Inhibidores tisulares de las metaloproteinasas
Se han descrito 4 inhibidores tisulares de las metaloproteinasas (TIMP). Aunque todos pueden inhibir a las MMP, su afinidad es distinta para cada una de ellas. El TIMP2 es el que ha demostrado tener mayor afinidad por MMP2 y MMP9. En el estudio realizado por Hirano et al26 se encontraron 2 variantes génicas del TIMP2. En una había una sustitución guanina/adenosina en la posición +853 en el exón 3 y en la otra una sustitución guanina/citosina en la posición 418 de la región del promotor. El primer polimorfismo no supone un cambio de aminoácido, pero es posible que influya en la estructura secundaria del ARNm e interfiera en su estabilidad. El polimorfismo de la región del promotor conlleva una menor actividad transcripcional del gen TIMP2. Los autores encontraron una frecuencia significativamente mayor del alelo +853 en el grupo de pacientes con EPOC comparado con el grupo control. El alelo 418 también presentaba una tendencia a ser más frecuente en el grupo de los sujetos con enfermedad pulmonar.
Alfa-1 antiquimiotripsina
Al igual que la AAT, la alfa-1 antiquimiotripsina es una proteína inhibidora de las serinproteasas, que actúa principalmente en la quimiotripsina pancreática, la catepsina G del neutrófilo y la quimasa mastocitaria. Además, disminuye la producción de superóxido por parte del neutrófilo22. Es sintetizada por los hepatocitos y los macrófagos alveolares. Se han encontrado 3 mutaciones en el gen de la alfa-1 antiquimiotripsina, que presenta un patrón de herencia autosómico dominante. Su déficit no se ha asociado a ninguna enfermedad, aunque se ha encontrado una mayor prevalencia en pacientes con EPOC y en el asma infantil. La sustitución Pro227-Ala se ha relacionado con la EPOC en un estudio realizado en Alemania, ya que se encontró en 4 de los 100 casos de EPOC y en ninguno de los 100 sujetos sanos (p = 0,04). La mutación Leu55-Pro también se encontró con una frecuencia mayor en el grupo de los pacientes con EPOC. Sin embargo, esta asociación no se ha encontrado en otros estudios similares, por lo que no hay datos concluyentes16,22.
Estrés oxidativo
El desequilibrio entre oxidantes exógenos y antioxidantes endógenos produce la lesión de los componentes de la matriz extracelular y del epitelio de la vía aérea, así como inflamación mediada por citocinas. El humo del tabaco contribuye al estrés oxidativo, ya que es una gran fuente de oxidantes, principalmente radicales libres y óxido nítrico, y favorece la acumulación de leucocitos en el pulmón, que también liberan radicales libres. Por tanto, es de suponer que cualquier alteración genética en las enzimas antioxidantes presentes en el pulmón, como la glutatión-S-transferasa (GST), la superóxido dismutasa (SOD) y la catalasa, entre otras, produzca un aumento del estrés oxidativo y, por tanto, del daño pulmonar producido por el tabaco16.
Glutatión-S-transferasa
Las GST constituyen una familia de enzimas que participan en la destoxificación de sustancias electrofílicas y productos del estrés oxidativo derivados del humo del tabaco18,21. Pueden actuar tanto en el citosol como en la membrana celular. Se han identificado más de 20 genes distintos de GST, y algunos de sus polimorfismos se han relacionado con la EPOC.
GSTP1 se expresa abundantemente en los alvéolos, los macrófagos alveolares y los bronquiolos respiratorios. Se conocen 2 polimorfismos, aunque sólo uno modifica la actividad catalítica de la enzima22. La sustitución de isoleucina por valina en la posición 105 puede alterar la actividad de la enzima debido a que esta posición está muy cerca del sitio de fijación de la enzima a los sustratos sobre los que actúa. En un estudio realizado en Japón, se encontró una proporción significativamente mayor de individuos homocigotos para este alelo entre los pacientes con EPOC que en los controles sanos18. Sin embargo, en el estudio SAPALDIA, recientemente publicado, no se observó ninguna asociación entre GSTP1 y ninguno de los parámetros de función pulmonar evaluados27.
GSTM1 se expresa en las mismas células que la anterior, y al igual que ésta tampoco se ha encontrado relación entre este genotipo y alguna alteración de la función respiratoria27.
En cambio, los resultados de este estudio indican que el déficit genético de GSTT1, aislado o en combinación con la deficiencia de GSTM1, está asociado de manera independiente a una caída de la función pulmonar, en relación con la edad, más rápida en los varones que en las mujeres. Esta asociación es independiente del hábito tabáquico. Los varones homocigotos para la delección de GSTT1 presentaban un exceso de deterioro anual del FEV1 de 5,3 ml/año (p = 0,001)27.
Superóxido dismutasa
Existen 3 tipos de SOD. La SOD extracelular (SOD3) está presente en altas concentraciones en el pulmón, principalmente alrededor de los bronquios y junto a los alvéolos, donde hay gran cantidad de colágeno tipo 1. Se ha descrito un polimorfismo de SOD3 que aumenta los valores plasmáticos de la enzima. En varios estudios se ha constatado que la presencia de esta mutación se asocia a un riesgo menor de desarrollar EPOC incluso en los sujetos fumadores, por lo que se ha atribuido un papel protector del pulmón a esta proteína16.
Hidrolasa epóxido microsomial
La hidrolasa epóxido microsomial se expresa en varias células, entre ellas las epiteliales bronquiales. Esta enzima actúa en el pulmón, metabolizando los epóxidos intermediarios altamente reactivos que se forman en el humo del tabaco y que producen daño pulmonar. Existen 2 polimorfismos que alteran la actividad enzimática. Uno, en el exón 3, es una sustitución de histamina por tirosina en posición 113, y el otro, en el exón 4, se produce por sustitución de arginina por histamina en posición 139. En ambos casos, la variante con histamina se asocia a unos valores más bajos de actividad de la enzima y se ha relacionado con una mayor prevalencia en pacientes con enfisema y EPOC que en sujetos sanos16,18. No obstante, otros estudios han obtenido resultados distintos.
Hemooxigenanasa 1
La hemooxigenasa 1 (HMOX1) es una enzima catalizadora de la oxidación del grupo hemo y contribuye a la formación de antioxidantes. Se encuentra en mayor cantidad en el pulmón de los individuos fumadores que en el de los no fumadores, lo que indica una regulación positiva de su expresión en respuesta a los radicales libres del tabaco. La presencia de un microsatélite GTn repetido en la región 5' de la HMOX1 parece alterar la transcripción del gen en situación de estrés. La presencia de una repetición larga, más de 30 repeticiones, dificulta la inducción de la transcripción por los radicales libres, por lo que su presencia en sujetos fumadores les haría más susceptibles de desarrollar enfisema, al no poder proteger sus pulmones del daño oxidativo producido por el tabaco. Aunque hay algún trabajo que confirma esa hipótesis, son necesarios más estudios para confirmar los resultados16,18.
Citocromo P4501A1
El citocromo P4501A1 se localiza en todo el pulmón y se ha relacionado con la activación de procarcinógenos. La mutación en el exón 7 se ha relacionado con una mayor predisposición a desarrollar enfisema centroacinar, en pacientes con cáncer de pulmón18.
TNF-α
El TNF-α es una citocina proinflamatoria de la que se han descrito varios polimorfismos. Su papel en la patogenia de la EPOC no está definido. Existen estudios en la población tailandesa y japonesa que han encontrado una asociación entre el alelo TNF-α-308A y la EPOC, pero otros trabajos realizados en población caucásica no han confirmado esos resultados18.
TGF-β
El TGF-β1 regula la producción de matriz extracelular, el crecimiento y la diferenciación celular, y la reparación tisular. Los ratones que no pueden activar el TGF-β latente desarrollan enfisema por alteraciones de la MMP12. Se han realizado algunos trabajos que muestran una asociación entre algunos polimorfismos génicos del TGF-β1 y la EPOC aunque son necesarios más estudios para confirmar estos resultados16.
Interleucina 13
La sobreexpresión de IL-13 por células T-helper 2 produce inflamación y destrucción pulmonar mediada por metaloproteinasas y cisteininproteasas. Estudios experimentales en ratones han mostrado que la producción aumentada de IL-13 da lugar a enfisema con metaplasia mucosa. Se ha relacionado un polimorfismo localizado en la región del promotor con un aumento de la expresión de IL-1316.
Correspondencia:
Dra. E. Antón Díaz.
Hospital de La Princesa, C/ Diego de León, 62. 28006. Madrid. España.
Correo electrónico: eantondiaz@telefonica.net







