


Introducción
El síndrome de Cowden es una enfermedad rara, que sigue un modelo de herencia dominante y cuya manifestación más frecuente es en forma de tumores hamartomatosos benignos, principalmente pólipos colónicos, por lo que esta enfermedad se clasifica dentro de las poliposis colónicas. Este síndrome está relacionado con mutaciones en la línea germinal del gen PTEN, que favorecen el desarrollo de tumores tanto malignos como benignos de la piel, tiroides, mama y cerebro.
El hemangioma esclerosante de pulmón es una neoplasia pulmonar poco frecuente, cuya primera descripción realizaron Liebow y Hubbel1 en 1983. En la actualidad su histología está bien definida, pero su histiogénesis y su tratamiento están en discusión. Se reconocen 4 patrones histológicos: papilar, sólido, esclerótico y hemorrágico2-4. Uno de los principales problemas estriba en diferenciar el hemangioma esclerosante del carcinoma pulmonar con patrón papilar5.
La fisiopatología de la asociación entre el síndrome de Cowden y el hemangioma esclerosante no está clara, aunque en la literatura médica se han descrito casos de aparición conjunta de hemangioma esclerosante de pulmón y otras poliposis, lo que podría indicar que el hemangioma esclerosante podría ser una más de las malformaciones posibles presentes en el síndrome de Cowden.
Observación clínica
Mujer de 18 años de edad, remitida para el estudio de un nódulo pulmonar solitario que se había hallado de forma casual en una radiografía de control. Entre los antecedentes personales destacaban hiperplasia nodular múltiple de tiroides, malformación vascular venosa de alto flujo en el tercio distal del muslo derecho y en la espalda, que se habían extirpado quirúrgicamente, lipomas perirrenales, retroauriculares y supraclaviculares intervenidos. Además, se le había diagnosticado de quiste ovárico y de fibroadenomas mamarios. En una radiografía simple de tórax de control realizada durante el seguimiento endocrinológico se había observado un nódulo pulmonar solitario, de aproximadamente 2 cm, en el lóbulo superior derecho, por lo que se la envió al servicio de neumología para estudio.
La paciente no presentaba antecedentes de hábitos tóxicos ni alergias a medicamentos. No describía fiebre, tos ni expectoración, astenia, anorexia ni pérdida de peso. En la exploración física no se objetivaron adenomegalias y la auscultación cardiopulmonar fue normal, al igual que los datos analíticos (hemograma, bioquímica y coagulación), salvo la presencia de anemia. Se realizó estudio inmunológico y de marcadores tumorales, cuyos valores se encontraban dentro de la normalidad. Ante el hallazgo del nódulo pulmonar solitario se realizó una fibrobroncoscopia con aspirado bronquial y lavado broncoalveolar, que fueron negativos para células tumorales e inflamatorias. La tomografía axial computarizada (TAC) mostró un nódulo sólido en el lóbulo superior derecho, de aproximadamente 2 cm y contornos lobulados, con una dudosa imagen de cavitación periférica, que no se realzaba con el contraste intravenoso. No se observaron adenopatías en rango significativo. En una TAC de control efectuada a los 3 meses la lesión no había cambiado de tamaño ni de morfología (fig. 1).
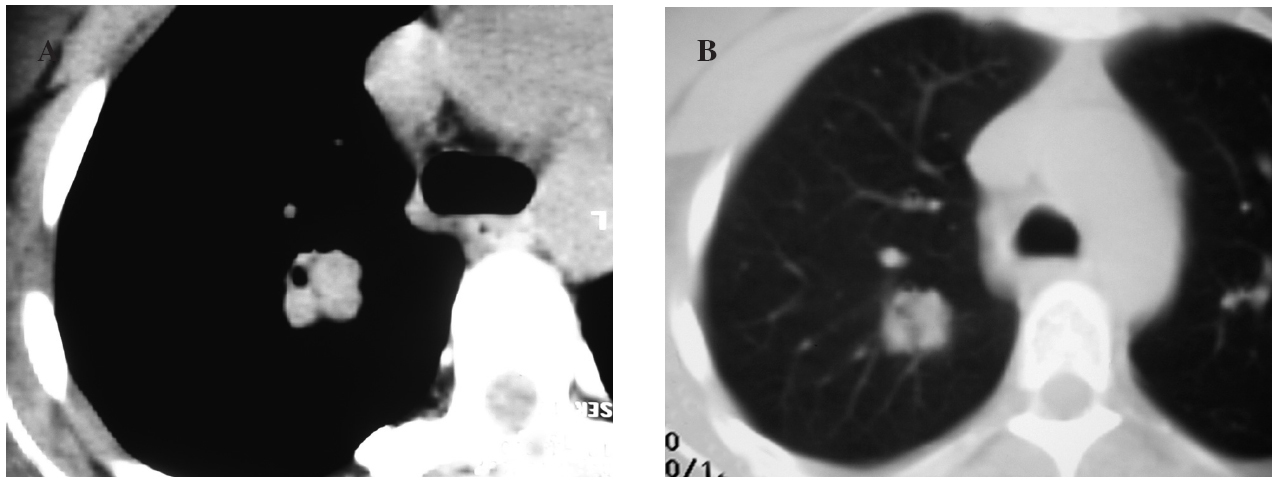
Fig. 1. Tomografía computarizada con contraste intravenoso en el cayado aórtico. A: ventana de mediastino, donde se observa una lesión sólida y polilobulada en el lóbulo superior derecho, de densidad de partes blandas y seudocavitada, que no realza con el contraste intravenoso. B: ventana de parénquima pulmonar, donde la lesión está bien definida y es más homogénea.
Se realizó punción aspirativa con aguja fina bajo control de TAC del nódulo pulmonar, con el resultado de proliferación epitelial de morfología papilar focal y atipia aislada, pero sin signos de malignidad. Dado que el resultado no era concluyente, se realizó una toracotomía derecha con biopsia del nódulo (fig. 2), cuyo resultado fue de hemangioma esclerosante. Dadas la edad y la pluripatología de la paciente, se pidió un estudio genético, que resultó ser indicativo de síndrome de Cowden, por lo que se decidió remitirla al servicio de digestivo para descartar la presencia de pólipos hamartomatosos colónicos.
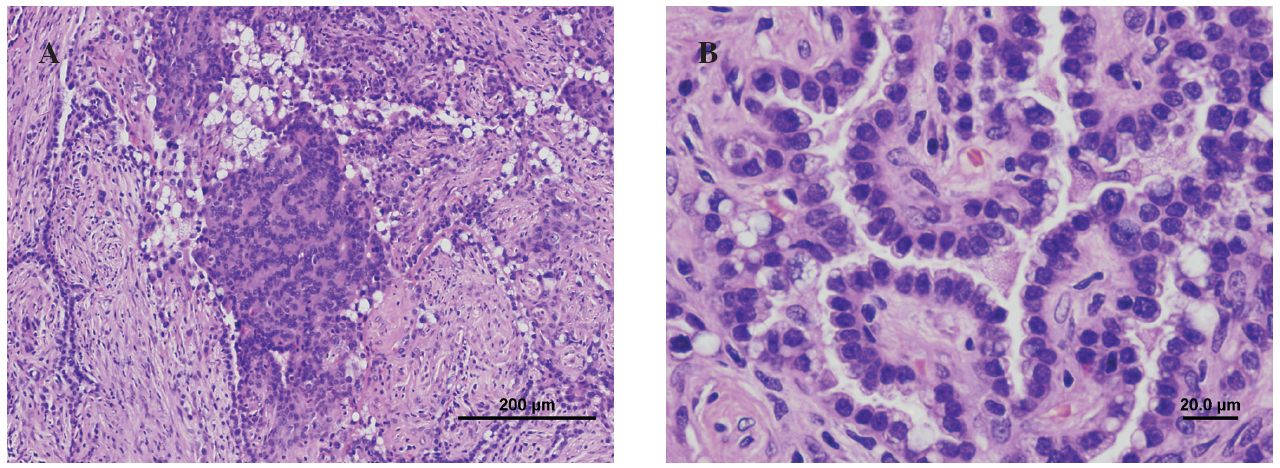
Fig. 2. Anatomía patológica. A: formación nodular mal delimitada, con una arquitectura predominantemente papilar, salpicadas áreas sólidas y escasas áreas esclerosadas. B: proliferación de células monomorfas, de forma poligonal, con núcleos grandes de bordes irregulares y cromatina grumosa. Los citoplasmas, que presentan frecuentes vacuolas apicales, son densos, eosinófilos y de aspecto sincitial en las áreas sólidas. En las zonas papilares las células se disponen como una monocapa que tapiza los ejes conjuntivos anchos con abundantes células fibroblásticas.
Actualmente la paciente continúa en seguimiento, sin precisar ningún tratamiento específico, y no ha presentado nuevas complicaciones relacionadas con su enfermedad de base.
Discusión
El hemangioma esclerosante es un tumor raro, que ha recibido numerosos nombres, tales como neumocitoma esclerosante benigno, histiocitoma pulmonar benigno, angioma esclerosante y seudotumor xantomatoso6. Descrito inicialmente como un tumor vascular con infiltración celular, zonas de esclerosis y hemorragia1,7, su origen histológico ha sido ampliamente discutido. Se le ha asignado un origen mesotelial, del mesénquima alveolar indiferenciado y del epitelio alveolar, pero los estudios de inmunohistoquímica y microscopia electrónica han hecho que actualmente sean los neumocitos de tipo II las células más aceptadas en sus histogénesis2,4,8. En la inmunohistoquímica se caracteriza por la positividad de los marcadores TTF-1 (factor de transcripción tiroidea-1) y del antígeno epitelial de membrana tanto en las células redondas como en las de superficie.
Más del 95% de los pacientes son mujeres (5:1), con un rango de edad comprendido entre los 6 y 83 años (media: 46 años), aunque se han descrito casos en pacientes pediátricos menores de 6 años. La mayoría de las veces es un hallazgo casual (70-80%), como en el caso que presentamos.
En la radiografía de tórax la lesión aparece como un nódulo solitario bien delimitado, con un tamaño variable, que oscila entre 0,3 y 7 cm, con un promedio de 2,6 cm, siendo el 73% de las lesiones menores a 3 cm, datos que coinciden con nuestro caso. Su localización suele ser subpleural, preferentemente en los lóbulos inferiores, con un discreto predominio en el pulmón derecho, aunque en nuestra paciente el nódulo se localizó en el lóbulo superior derecho y central (fig. 1). En la TAC se presenta como una masa bien definida subpleural, con realce de contraste. Estos últimos datos difieren de los presentados en la TAC de tórax con contraste de nuestro caso, dado que se visualizaba un único nódulo sólido, de contornos bien definidos, de unos 2 cm de diámetro, difícil de percibir con la ventana de mediastino, sin realce de contraste, de situación central en el lóbulo superior derecho. Además, no se había modificado en un control posterior, a los 3 meses.
En el estudio macroscópico se encontró una lesión de bordes definidos, no encapsulada, que variaba de una tonalidad gris-blanquecina a amarillenta y rojo oscuro, de consistencia sólido-quística, con áreas esponjosas. Histológicamente se caracterizaba por estar compuesta por células redondas, con estructuras papilares y tubulares entremezcladas con zonas escleróticas y angiomatosas. La mayoría de los hemangiomas esclerosantes presentan un patrón papilar. En el estudio de Katzenstein et al5, el patrón sólido estaba presente en todos los pacientes (n = 51); el hemorrágico, en 37; el papilar, en 38, y el esclerótico fue más evidente en 50 casos. El diagnóstico de carcinoma papilar de tiroides metastásico, mesotelioma y adenocarcinoma bronquioloalveolar puede considerarse en los casos en que predomina el componente papilar.
El síndrome de Cowden es una enfermedad autosómica dominante, que se caracteriza por múltiples hamartomas derivados del endodermo, mesodermo y ectodermo. Está causado por mutaciones en la línea germinal del gen PTEN (10q23), que parece estar en relación con procesos de señalización y adherencia celulares. No obstante, hay otras enfermedades donde también existe mutación del gen PTEN, como el síndrome de Bannayan-Ruvalcaba-Riley (recientemente descrito como variante del síndrome de Cowden), la enfermedad de Lhermitte-Duclos y un subgrupo de poliposis familiar juvenil9,10, así como en múltiples tumores esporádicos.
Clásicamente el síndrome de Cowden se ha clasificado dentro de los síndromes de poliposis intestinales hamartomatosos. Este síndrome asocia lesiones verrugosas de la piel, tricolemomas, gangliocitoma cerebeloso y adenomas tiroideos, así como hemangiomas cutáneos, malformaciones arteriovenosas viscerales, enfermedad fibroquística y cáncer de mama9,10, muchos de los cuales estaban presentes en nuestra paciente.
Aunque no se ha descrito la asociación del hemangioma esclerosante pulmonar y el síndrome de Cowden, la literatura médica recoge casos de hemangioma esclerosante pulmonar con poliposis adenomatosa familiar, probablemente por expresión aberrante de la betacatenina, un oncogén activador de la transcripción del Wnt, que está regulado por la proteína APC11. Asimismo, se ha descrito la asociación del hemangioma esclerosante de pulmón a miomas, quistes de tiroides y riñón12.
Éste es el primer caso descrito de asociación entre el hemangioma esclerosante pulmonar y el síndrome de Cowden, por lo que, dada la multitud de lesiones asociadas a este síndrome, cabría pensar que el hemangioma esclerosante podría ser una más de estas alteraciones. Sin embargo, dada la escasa prevalencia de las 2 enfermedades, no se ha podido profundizar en la posible asociación entre ambas. Este caso pone de manifiesto la heterogeneidad de las lesiones asociadas al síndrome de Cowden y que, aunque algunas de ellas sean poco prevalentes, como es el caso del hemangioma esclerosante de pulmón, hay que considerarlo dentro del diagnóstico diferencial del nódulo pulmonar solitario en estos pacientes.
Correspondencia: Dr. F. Guerra-Gutiérrez.
Servicio de Radiología. Hospital Universitario La Paz.
P.o de la Castellana, 261. 28046 Madrid. España.
Correo electrónico: flaguers@hotmail.com
Recibido: 17-4-2006; aceptado para su publicación: 18-7-2006.







