





Idiopathic pulmonary fibrosis (IPF) is progressive and irreversible. Some discrepancies about IPF staging exists, especially in mild phases. Forced vital capacity (FVC) higher than 80% has been considered early or mild IPF even for the design of clinical trials.
MethodsSpanish multicentre, observational, retrospective study of IPF patients diagnosed between 2012 and 2016, based on the ATS/ERS criteria, which presented FVC greater or equal 80% at diagnosis. Clinical and demographic characteristics, lung function, radiological pattern, treatment, and follow-up were analyzed.
Results225 IPF patients were included, 72.9% were men. The mean age was 69.5 years. The predominant high-resolution computed tomography (HRCT) pattern was consistent usual interstitial pneumonia (UIP) (51.6%). 84.7% of patients presented respiratory symptoms (exertional dyspnea and/or cough) and 33.33% showed oxygen desaturation below 90% in the 6min walking test (6MWT). Anti-fibrotic treatment was initiated at diagnosis in 55.11% of patients. Median FVC was 89.6% (IQR 17) and 58.7% of patients had a decrease of diffusion lung capacity for carbon monoxide (DLCO) below 60% of theoretical value; most of them presented functional progression (61.4%) and higher mortality at 3 years (20.45%). A statistically significant correlation with the 3-years mortality was observed between DLCO <60% and consistent UIP radiological pattern.
ConclusionsPatients with preserved FVC but presenting UIP radiological pattern and moderate–severe DLCO decrease at diagnosis associate an increased risk of progression, death or lung transplantation. Therefore, in these cases, preserved FVC would not be representative of early or mild IPF.
Introducción La fibrosis pulmonar idiopática (FPI) es progresiva e irreversible. Existen algunas discrepancias sobre la estadificación de la FPI, especialmente en las fases leves. La capacidad vital forzada (FVC) superior al 80% se ha considerado una FPI temprana o leve incluso para el diseño de ensayos clínicos.
MétodosEstudio español multicéntrico, observacional, retrospectivo de pacientes con FPI diagnosticados entre 2012-2016, en función de los criterios ATS/ERS, que presentaban FVC mayor o igual al 80% al diagnóstico. Se analizaron características clínicas y demográficas, función pulmonar, patrón radiológico, tratamiento y seguimiento.
ResultadosSe incluyeron 225 pacientes con FPI, el 72,9% eran varones. La edad media fue de 69,5 años. El patrón predominante en la tomografía computarizada de alta resolución (TCAR) fue compatible con neumonía intersticial usual (NIU) (51,6%). El 84,7% de los pacientes presentó síntomas respiratorios (disnea de esfuerzo o tos) y el 33,33% mostró desaturación con una saturación de oxígeno inferior al 90% en la prueba de la marcha de los 6min (PM6M). El tratamiento antifibrótico se inició en el momento del diagnóstico en el 55,11% de los pacientes. La mediana de la CVF fue del 89,6% (RIC 17) y el 58,7% de los pacientes presentó una disminución de la capacidad pulmonar de difusión del monóxido de carbono (DLCO) por debajo del 60% del valor teórico; la mayoría presentó progresión funcional (61,4%) y una mayor mortalidad a los 3 años (20,45%). Se observó una correlación estadísticamente significativa de la mortalidad a los 3 años entre una DLCO<60% y el patrón radiológico compatible con UIP.
ConclusionesLos pacientes con FVC conservada pero que presentan patrón radiológico de NIU y disminución moderada-grave de la de DLCO en el momento del diagnóstico se asociaron a un mayor riesgo de progresión, fallecimiento o tener que recibir un trasplante de pulmón. Por lo tanto, en estos casos, una FVC preservada no sería representativa de una FPI temprana o leve.
Idiopathic pulmonary fibrosis is a progressive, irreversible, chronic and lethal interstitial lung disease (ILD).1 Natural history differs greatly between patients but involves a decline in lung function that negatively impacts quality of life.2 The disease course of IPF is highly variable; most patients progress more slowly while others experience rapid lung decline. In addition, may have periods of relatively stable disease interspersed with acute deteriorations in lung function.3 With the advent of pharmacological and non-pharmacological treatment options that slow disease progression and try to preserve quality of life, it is more important than ever for IPF patients to be diagnosed quickly and given access to the required holistic care.4–7
The delay in diagnosis has become a factor associated with increased mortality.8,9 The time from the onset of symptoms to diagnosis ranges from 9 months to 3 years depending on the region where the patient lives. Nevertheless, the proportion of cases with preserved FVC has increased in the recent years due to various factors such as training programs for early identifying ILD, higher awareness about the disease, and better radiological recognition and follow-up of interstitial lung abnormalities.10–12 However, these patients present wide variability in the progression, dependent on clinical factors and associated comorbidities.9,11 Anti-fibrotic treatment initiation has been delayed in IPF patients with preserved FVC in different countries,12–15 although some studies demonstrated the same benefit for these cases.16,17 A recent international survey demonstrates that among the reasons for the “wait-and-see” option after IPF diagnosis the preserved FVC is predominant.15,18 Probably, it is in part due to the fact that presenting FVC above 80% is usually associated with the idea of “early” or “mild” disease.12 Therefore, identifying the different IPF patient profiles among those cases with preserved FVC would be of interest for optimizing therapeutic approach from diagnosis.
The present study aims to evaluate the clinical features and the 3 year-mortality predictive factors of IPF patients that present preserved FVC at diagnosis.
MethodsPopulation and study designMulticentre, observational and retrospective study that includes IPF patients diagnosed between 2012 and 2016 that presented FVC greater than or equal to 80% of its theoretical value. The included patients were followed-up every 6 months (clinical and pulmonary functional test evaluation). Patients were recruited from 18 Spanish hospitals that covered the different regions of the whole country, most of them were part of the Spanish IPF Registry or the Observatory IPF.cat. IPF diagnosis was evaluated in a multidisciplinary discussion including expert ILD physicians, pathologists and radiologists on the basis of the respective ATS/ERS/JRS/ALAT guidelines.1 HRCT scan images of all patients were checked by expert ILD radiologists following the same international consensus criteria ATS/ERS and Fleischner Society (only two centers didn’t work with expert ILD radiologist and they sent the anonymized HRCT images to the Multidisciplinary ILD Committee of University Hospital of Bellvitge). All patients provided written informed consent and the study was approved by the Institutional Ethics Committee (CEIC, ref. PR307/16). Data management has followed the regulatory bases included in the EU 2016/679 statement and the Declaration of Helsinki (Fortaleza, 2013). The follow-up was documented for 3 years.
Recruited dataBaseline demographic and clinical characteristics at diagnosis were collected, including age, gender, smoking history, comorbidities, respiratory symptoms (dyspnea, cough) and initiated treatment. Lung function variables (FVC, DLCO, oxygen desaturation in 6MWT) and radiological high-resolution computing tomography (HRCT) pattern at diagnosis were recorded. The HRCT findings and radiological pattern were interpreted on the basis of recommendations from the nomenclature committee of the Fleischner Society.19 Chest HRCT scans were performed on multidetector scanners, volumetric images being acquired with the patient lying in supine position, after maximal inspiration, and without iodinated contrast medium.
The characteristics for each identifiable radiological pattern in IPF were previously defined to improve not only the reading by each radiologist, but also to increase agreement among evaluations (Supplementary Table 1). The presence of honeycombing in those cases with consistent UIP pattern was graded (greater than or equal to 5%). Emphysema was defined as low-attenuation areas bordered by a very thin (<1mm) or no wall in the normal lung. Combined pulmonary fibrosis emphysema syndrome (CPFE) was considered when more than 15% was present.20,21
The annual follow-up data was collected until the end of the study, death, or lung transplantation. FVC and DLCO changes, appearance of new comorbidities such as lung cancer, acute exacerbations (characterized by rapid progression of dyspnea within the previous 30 days, with the presence of new bilateral ground-glass opacities or consolidation by radiography/HRCT without pneumo-thorax or pleural effusion, and a marked decrease in the PaO2),3 lung transplantation and mortality were documented. Disease progression was considered in cases that presented absolute decline of FVC≥10% and/or DLCO≥15% of theoretical value over one year.
Statistical analysisCategorical data are described as frequency and percentages, and continuous variables are expressed as mean±standard deviation (SD) or median±interquartile range as appropriate. Group comparisons were made using Chi-squared test or Fisher's exact test when required. Differences in continuous variables were analyzed with ANOVA tests or Student's t test, or their corresponding non-parametrical tests when required (Kruskal–Wallis and Mann–Whitney U tests). Time to event data (time to lung transplant and/or death) were analyzed using Kaplan–Meier analysis. Cox proportional hazard regression for multivariable analyses was used to determine whether the following factors at diagnosis increased the mortality risk: gender, age, smoking history, emphysema, DLCO, exercise capacity, radiological HRCT pattern (model Log-Rank test p-value <0.001; concordance index 0.8). A p-value <0.05 was considered statistically significant. Data were analyzed by using SPSS 24 (SPSS, IBM Corp) and R (software version 3.6.2).
ResultsDiagnostic IPF patient features (Table 1)From 225 patients, the majority were male (72.9%), had a mean age of 69.5 years, and 66.5% of them reported exposure to tobacco. The predominant radiological pattern in the HRCT was consistent UIP (51.6%), with an extension of more than 5% of honeycombing in 36.3% of them. Emphysema was present in 33.3% of cases, but only 12.9% fulfilled CPFE criteria. The median FVC was 89.6% (IQR 17) and DLCO was 56.2% (SD 18.5%). Interestingly, 58.7% of patients presented a decrease in DLCO<60% of predicted value and 44.7% of them had associated emphysema.
IPF patient features at diagnosis.
| Patients (n=225) | |
|---|---|
| Age, median (IQR) | 69.5 (12) |
| Gender (male), n (%) | 164 (72.9%) |
| Exposure to tobacco, n (%) | |
| Former | 112 (62.6%) |
| Current smokers | 9 (4%) |
| Respiratory symptoms, n (%) | 189 (84.8%) |
| FVC percentage of predicted, median (IQR) | 89.6 (17) |
| DLCO percentage of predicted, mean (SD) | 56.26 (18.5) |
| Chest HRCT pattern, n (%) | |
| Consistent UIP | 116 (51.6%) |
| Probable UIP | 86 (38.2%) |
| Associated emphysema, n (%) | 75 (33.3%) |
| CPFE, n (%) | 29 (12.9%) |
| Biopsy, n (%) | |
| Surgical biopsy | 73 (32.4%) |
| Cryobiopsy | 8 (3.6%) |
| DLCO<60% of predicted, n (%) | 132 (58.7%) |
| Associated emphysema | 59 (44.7%) |
| Functional progression | 81 (61.4%) |
| 3-year mortality | 27 (20.4%) |
| Oxygen saturation below 90% (6MWT), n (%) | 75 (33.3%) |
| Anti-fibrotic initiation at diagnosis, n (%) | 124 (55.1%) |
| Death or lung transplant at 3 years, n (%) | 33 (14.7%) |
FVC: forced vital capacity; DLCO: diffusing capacity for carbon monoxide; HRCT: high resolution computed tomography; UIP: usual interstitial pneumonia; 6MWT: 6-minute walking test; IQR: interquartile range; SD: standard deviation, CPFE (combined pulmonary fibrosis and emphysema).
Only 34 patients (15.1%) were asymptomatic. Of those, 9 (26.5%) presented normal DLCO (>80% predicted value) and indeterminate or probable UIP pattern in HRCT, and only 1 (2.9%) had normal DLCO and consistent UIP pattern. Of the asymptomatic patients, 14 (41.2%) had decreased DLCO (<80% pred.) and indeterminate or probable UIP pattern, and 10 (29.4%) had decreased DLCO and consistent UIP pattern. One subjects from this latter group died during follow-up, but no events were observed in patients with decreased DLCO and indeterminate or probable UIP.
Regarding IPF treatment, 124 (55.1%) of patients received anti-fibrotic medication at diagnosis, 46 cases nintedanib and 59 pirfenidone (in 19 patients the type of anti-fibrotic drug was missing).
IPF progression in patients with preserved FVC53.3% of patients presented disease progression at the 3-years of follow-up; of them 14.7% died or received lung-transplant within 3 years after diagnosis. Furthermore, from this group only 7 patients underwent lung transplant, the other 26 cases died.
The cause of death was due to IPF disease progression in most cases (74.8%), lung cancer in 1% and to other reasons not related to the disease in 24.2%. Only 2% of patients presented an acute exacerbation that required hospitalization. When comparing the group who died within 3 years with those who were still alive, we found differences between the two groups (Table 2).
Differences between IPF patients with preserved FVC at diagnosis that died compared with those that were alive after 3 years of follow-up.
| Patients dead or transplanted at 3 years (n=33) | Patients alive at 3 years (n=192) | p | |
|---|---|---|---|
| Age (years), median (SD) | 70 (8) | 69 (13) | 0.558 |
| Gender (male), n (%) | 27 (81.8%) | 137 (74.1%) | 0.463 |
| Smoking history, n (%) | 20 (86.9%) | 101 (63.5%) | 0.046* |
| FVC percentage of predicted, mean (SD) | 90 (16) | 89.2 (17) | 0.836 |
| DLCO percentage of predicted, mean (SD) | 42.1 (15.9) | 58.72 (17.9) | <0.001* |
| Chest HRCT pattern, n (%) | |||
| Consistent UIP | 22 (91.7%) | 94 (54.3%) | |
| Probable UIP | 1 (4.2%) | 71 (41%) | <0.001* |
| Indeterminate UIP | 1 (4.2%) | 8 (4.6%) | |
| Honeycombing >5%, n (%) | 24 (72.7%) | 57 (29.8%) | <0.001* |
| Emphysema, n (%) | 16 (48.5%) | 59 (30.9%) | 0.075 |
| CPFE, n (%) | 15 (51,72%) | 14 (48,28) | 0.792 |
| Dyspnea, n (%) | 31 (93.9%) | 133 (70%) | 0.007* |
| Cough, n (%) | 23 (71.9%) | 90 (50%) | 0.036* |
| Desaturation on 6MWT below 90%, n (%) | 21 (70%) | 54 (30.5%) | <0.001* |
| No antifibrotic treatment, n (%) | 17 (51.5%) | 84 (43.7%) | 0.522 |
FVC: forced vital capacity; DLCO: diffusing capacity for carbon monoxide; HRCT: high resolution computed tomography; UIP: usual interstitial pneumonia; 6MWT: 6-minute walking test; IQR: interquartile range; SD: standard deviation.
The patients who died were more symptomatic at diagnosis, and the majority presented dyspnea (93.9%) and cough (71.8%). All patients with no symptoms, normal DLCO and indeterminate or probable UIP pattern at diagnosis were alive at 3 years.
It should be noted that the group of patients that died had a lower DLCO at diagnosis (mean 42.1%±16 vs. 58.7%±17.9; p<0.001). When analyzing the basal DLCO in three degrees of severity (mild: DLCO 80–60%, moderate: 60–40%, severe: <40%22), we observed that patients presenting severe DLCO decrease had a worse prognosis and higher risk of mortality and/or lung transplantation (Fig. 1). Up to 81.8% of patients that died presented moderate-severe decrease of DLCO. Patients with severe DLCO reduction at diagnosis presented disease progression in 61.4% cases and a 3-year mortality of 20.4%.
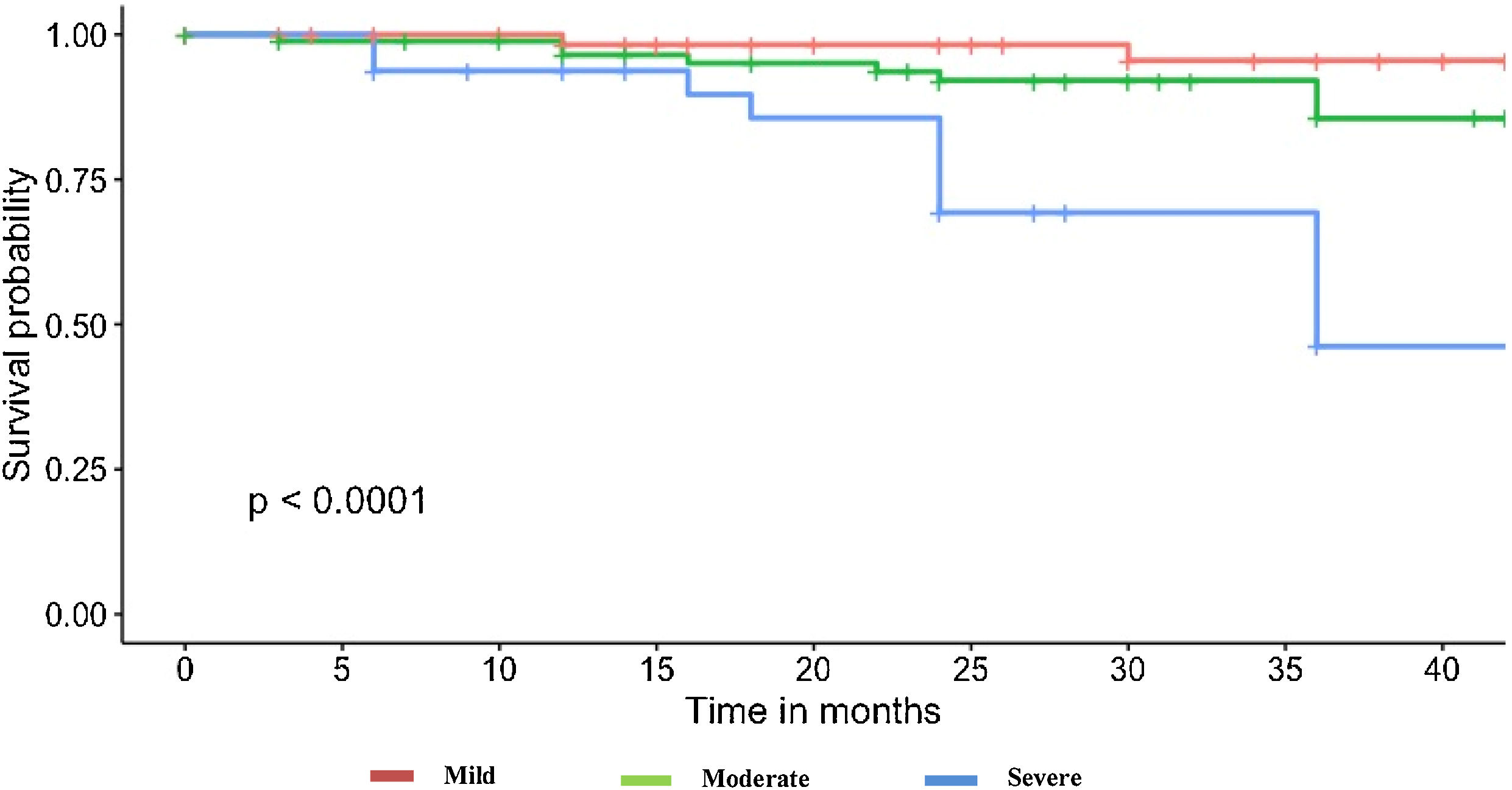
Oxygen desaturation below 90% during the 6MWT was also more frequently present at diagnosis among those cases that died (70%) than in the group that was alive (30.5%).
Regarding the chest HRCT pattern at diagnosis, we found remarkable differences. In the deceased group, the predominant pattern had been consistent UIP (91.7%). On the other hand, among those alive, the radiological patterns were more variable; 54.3% had a pattern of UIP, 41% probable UIP, and the pattern of indeterminate UIP was similar in both groups (4%). Among those cases with consistent UIP, the extension of honeycombing >5% was very frequent in the group of deaths (72.7%) when comparing with those that were alive (29.8%), p<0.001 (Fig. 2).
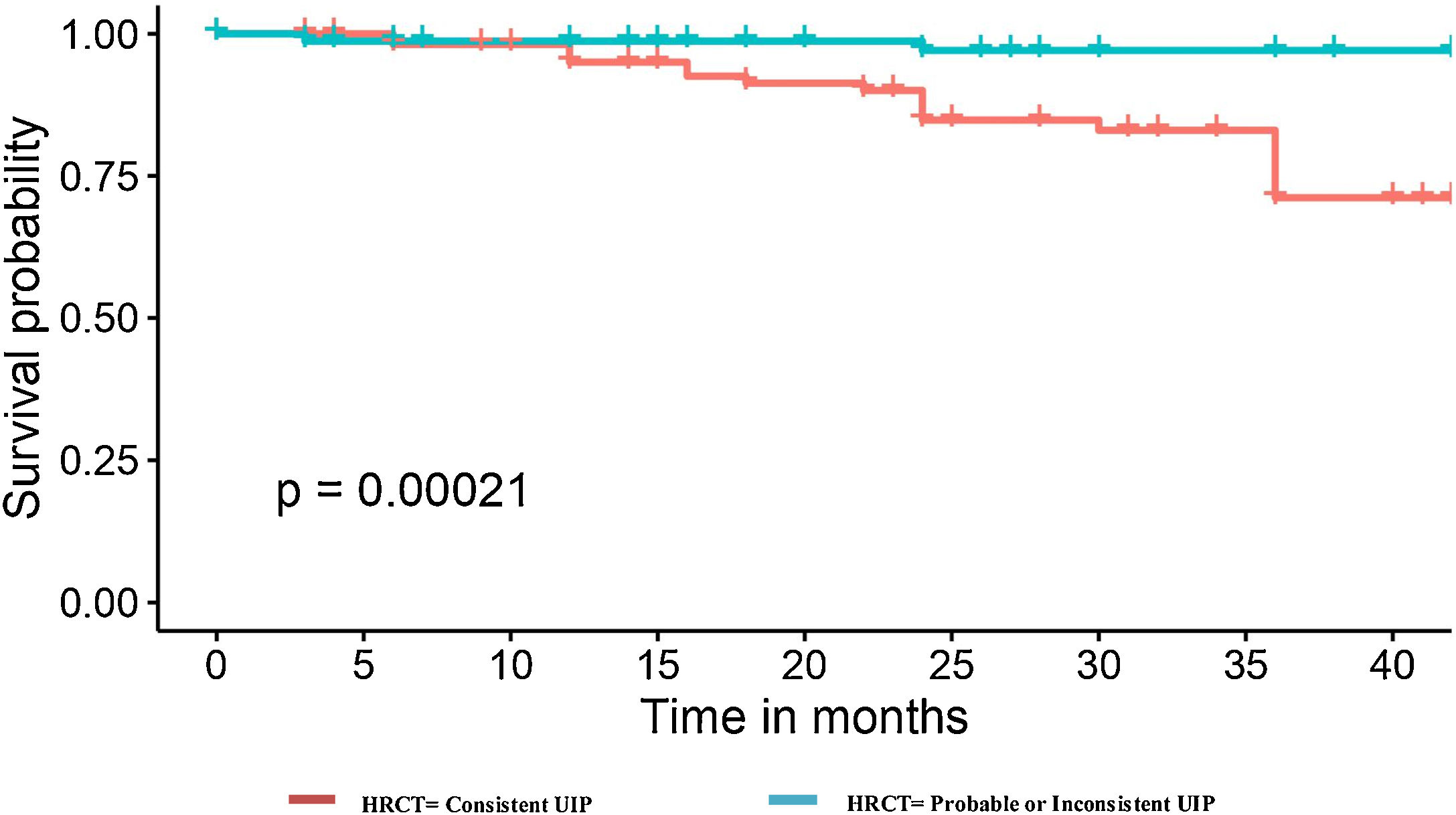
A higher proportion of former or current smokers was present in the group that died or underwent to lung transplant compared with the alive group after 3-years (86.9% vs. 63.5%, p=0.046). The presence of emphysema was not statistically different (0.075) and neither the CPFE cases. However, 15 of 16 IPF patients with emphysema that died met CPFE criteria and the mortality rate among CPFE cases was three times greater (51.7%) than in the global cohort (14.7%), which differs from those emphysema cases without CPFE criteria (2.17%). Therefore, the poor prognosis associated with smoking history could be attributed in part to those cases in which this habit induces CPFE.
Regarding anti-fibrotic medication, 48.5% of patients who were deceased at 3 years had received anti-fibrotic treatment from diagnosis, while 56.2% of patients that were alive had been treated from the beginning.
A Cox proportional hazard model was constructed with objective variables of clinical relevance and statistically significant on the univariate analyses, including age, gender, smoking habit, oxygen desaturation in the 6MWT, DLCO (categorized as mentioned above) and presence of UIP radiological pattern. The model showed that patients with a very severe DLCO decrease and consistent UIP radiological pattern had a higher risk for mortality (HR 7.4, 95% CI 1.06–51.9, p=0.043; and HR 5.2, 95% CI 1.13–23.5, p=0.034, respectively). Results for this model are shown in Fig. 3.
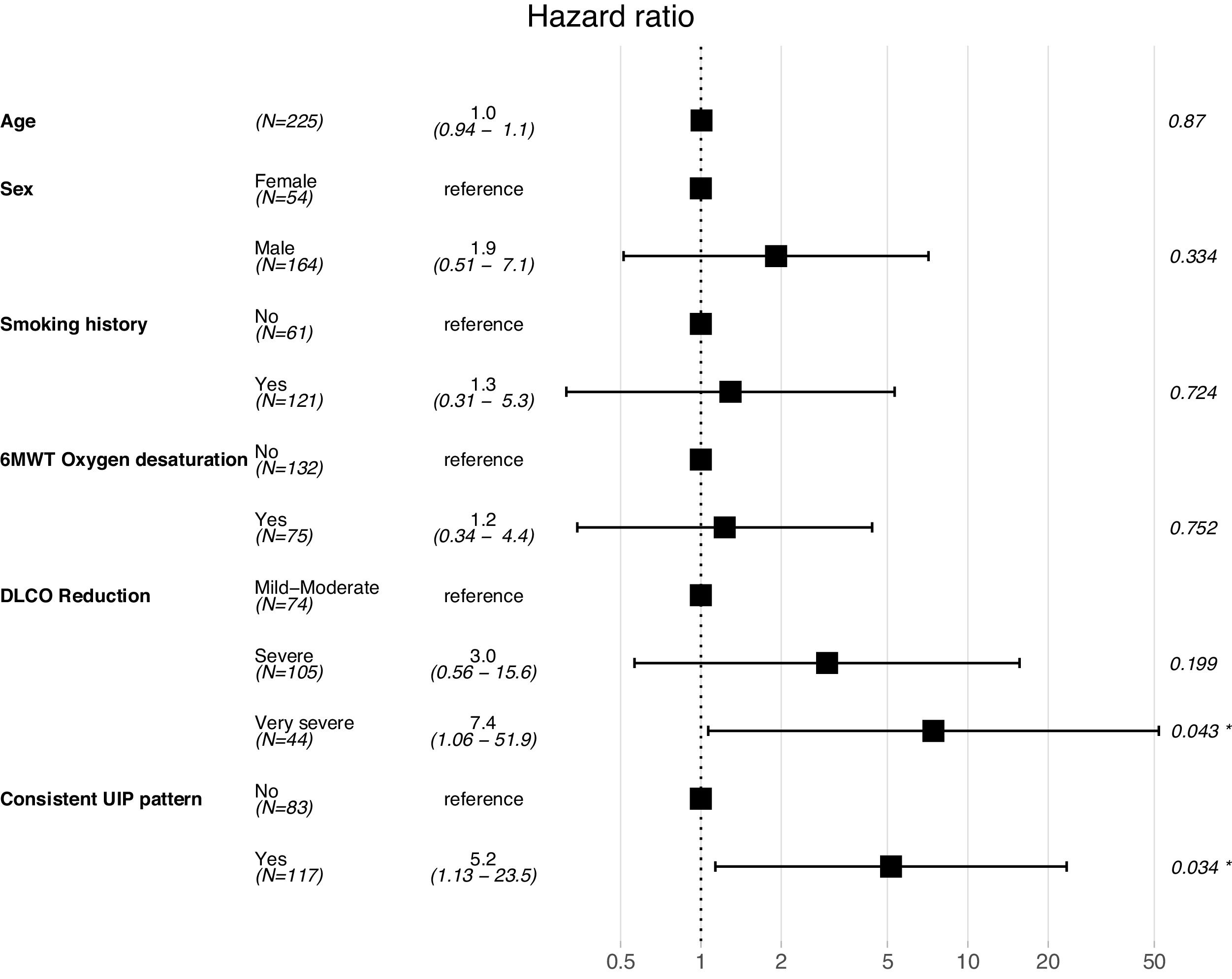
According to these results, the prognostic factors at diagnosis of IPF with preserved FVC were the presence of consistent UIP pattern in chest HRCT, the severe decrease in DLCO.
Staging IPF patients with preserved FVCConsidering IPF management depending on disease behavior, patients with preserved FVC at higher risk for 3-year mortality or lung transplant would not be considered early or mild IPF (including the presence of radiological UIP consistent pattern and CPFE criteria). Regarding the identified factors associated with disease behavior in these patients, we differentiated stage 0 or “early IPF” from the other IPF patient profiles with preserved FVC (Table 3). Stage 0 would include a minority of cases without symptoms that present with normal DLCO and indeterminate or probable UIP pattern in the chest HRCT. In our cohort, only 9 (4%) met these criteria. All of them were GAP stage I. Only one stage 0 patient showed disease progression, but none had died or received lung-transplant at 3 years of follow-up. Median FVC for stage 0 patients was 105.4% (IQR 18), and mean DLCO was 93% (SD 13.6%). Regarding chest HRCT pattern, six patients showed probable UIP, and 3 indeterminate UIP. Lung biopsy was required in 44.4% of these cases; 3 of them with morphological changes of consistent UIP and 1 probable UIP.
IPF patients with preserved FVC could be staged regarding other variables associated with prognosis, which would help in better managing these patients.
| Stage | HRCT pattern | SYMPTOMS (dyspnea and/or cough) | FVC (% pred.) | DLCO (% pred.) |
|---|---|---|---|---|
| 0 – “early” IPF | Truly indeterminate UIP (subpleural reticulation) or probable UIP | No | ≥80 | ≥80 |
| Non-zero | Probable or consistent UIP | Yes or no | ≥80 | <80 |
HRCT: high resolution computed tomography; FVC: forced vital capacity; DLCO: diffusing capacity for carbon monoxide; UIP: usual interstitial pneumonia.
On the other hand, non-early IPF patients would be characterized by normal FVC (≥80%) but decreased DLCO (<80%) and/or consistent or probable UIP and/or presence of respiratory symptoms (Table 3). Contrary to what was observed for stage 0, 119 (54.3%) subjects showed disease progression, and 33 (15%) died or required lung transplant at 3 years.
DiscussionAlthough more IPF patients with preserved FVC are being diagnosed in recent years, little is known about their features. However, only due to this functional respiratory condition many physicians have considered mild or early disease even for treatment decisions. Our study of a real-world clinical Spanish cohort demonstrates that IPF patients with preserved FVC are a heterogeneous group, but most of them present disease progression. The main risk factors for the 3-year mortality or lung transplant were a DLCO below 60%, HRCT pattern consistent UIP and the extension of honeycombing >5%. Considering disease behavior, only a minority of cases with preserved FVC could be considered “early IPF” at diagnosis. Therefore, staging IPF patients with preserved FVC regarding other prognostic factors may identify those cases at higher risk of disease progression and, consequently, lead to better optimizing treatment from diagnosis.
Prognostication in IPF is fraught with difficulties. An array of prognostic IPF features and indicators at diagnosis have been used over the years, but with varying degrees of success. Selman et al. identified a group of IPF patients with rapid progression and they found a significant correlation with age and smoking history.20 One reason for preserved FVC in patients with symptomatic IPF and severe DLCO deterioration is the presence of extended emphysema or the CPFE syndrome.21,23 Cottin et al. concluded that emphysema greater than or equal to 15% was associated with lower decrease in FVC over 48 weeks compared to less than 15% or no emphysema in IPF patients.21 12.9% of cases we evaluated met criteria for CPFE and half of them died over 3 years from diagnosis. On the other hand, only 2.17% of IPF cases with emphysema that didn’t meet CPFE criteria died at 3 years. Therefore, the poor prognosis associated with smoking history could be, in part, due to those cases that develop CPFE.
FVC and DLCO are the most sensitive markers of disease severity and prognosis.24,25 FVC decreases by an average of 150–250ml per year in untreated IPF patients.25,26 The GAP model (gender, age, FVC-DLCO) is a practical and easy means of determine the risk of death in patients with IPF.27-29 Ley et al.11,30 made reference to the advantage of adding the HRCT score to the GAP model (CT-GAP).
Currently, there is growing interest in the identification of early IPF. Regarding our results, early IPF would include a minority of patients with preserved FVC and DLCO, limited subpleural reticulation, for whom an indeterminate or probable UIP pattern is present and a lung biopsy is more frequently required. 9 patients from our IPF cohort met these clinical, radiological and functional criteria. Therefore, based on this description, these patients could be categorized as early IPF or interstitial lung abnormalities (ILAs).1,31,32 No consensus exists for the optimal management of this type of patients. Clinical associations with ILAs and IPF were strikingly similar. ILAs increased in prevalence with aging.31,32 ILA was present in approximately 7% of evaluated adult populations and the pattern of progression was variable but associated with increased risk of pulmonary function decline or death.32 Some studies demonstrated similarities in radiological, physiological, and genetic features between ILA and IPF, suggesting that some cases would represent an early stage of IPF.33
Our results show that only half of those IPF cases with preserved FVC that died had been treated with anti-fibrotic medication, although some studies show treatment effects have been similar for patients with FVC thresholds of above 80%.16,17 Recent studies suggest a reluctance to treat patients with ‘mild’ disease, and instead adopt a ‘watch and wait’ approach.9,15–17 According to the results of Maher et al.,12 40% of patients with a confirmed diagnosis of IPF, received no treatment. A higher proportion of untreated patients had FVC>80% compared to treated patients. Of the patients with “mild” IPF, 71% did not receive an approved antifibrotic versus 41% of patients with “moderate” IPF. Limited evidence exists about the natural history of IPF patients with normal FVC.17,34 A recent clinical trial brings relevant evidence about the anti-fibrotic benefits of nintedanib in patients with preserved FVC.17 In this clinical trial, most included patients presented respiratory symptoms or abnormal DLCO. Therefore, the minority of IPF patients with preserved FVC and “early IPF” have not been evaluated for anti-fibrotic medication.
Due to the retrospective nature of the included data, the results present several limitations. Data about comorbidities and complications such as pulmonary hypertension (PH) were not rigorously documented. However, it is probable that a high proportion of patients with severe deterioration of DLCO, especially those with CPFE, could present associated PH. Furthermore, no conclusion can be made regarding the effect of anti-fibrotic medication at diagnosis due to the study design and also because pirfenidone and nintedanib received the approval in 2014 and 2015 respectively in our country.
ConclusionPreserved FVC may reflect different types of IPF patients and is not enough to state early IPF. This observation associates prognostic and therapeutic implications when considering initiation of antifibrotic treatment or lung transplant evaluation. DLCO decrease or consistent UIP pattern at diagnosis are associated with a poor prognosis. Therefore, a better staging of IPF with preserved FVC by including these prognostic factors would aid in better managing these patients and optimizing treatment decisions.
FundingWe acknowledge:
- -
The unrestricted grant from Boeringher Ing. BI had no role in the design, analysis or interpretation of the results in this study. BI was given the opportunity to review the manuscript for medical and scientific accuracy as it relates to BI substances, as well as intellectual property considerations.
- -
The support of Roche to the SEPAR IPF Registry.
- -
The support and interaction with the Barcelona Respiratory Network (BRN) and Fundation Ramon Pla Armengol.
Maria Molina-Molina has received support as research grant and scientific advise from: Roche, Boehringer Ing, Pfizer, Galapagos, Origo Pharma, Esteve-Teijin, Chiesi.
The rest of authors declare that they have no conflict of interest.
The following are the supplementary data to this article:


☆ Supported by the Spanish Respiratory Society (SEPAR, PII EPID), Barcelona Respiratory Network (BRN), Ramón Plà Armengol Fundation, Boehringer Ingelheim and Hoffmann-La Roche.







