


Introducción
Entre un 7 y un 15% de las neoplasias se manifiestan en algún momento de su evolución con alguna forma de síndrome paraneoplásico. El porcentaje en el cáncer de pulmón llega a alcanzar al 10% de los pacientes, e incluye diferentes órganos y sistemas, de los que los más frecuentemente descritos son los neurológicos1 (síndromes miasteniformes, neuropatías, encefalitis o retinopatías) y endocrinológicos (síndrome de secreción inadecuada de hormona antidiurética, producción ectópica de corticotropina e hipercalcemia).
La incidencia del síndrome paraneoplásico cutáneo en el carcinoma broncogénico es baja2-5. En muchas ocasiones el reconocimiento y la valoración de los síntomas y signos que acompañan permiten sospechar el diagnóstico tumoral de manera precoz. Describimos un caso poco frecuente de síndrome paraneoplásico cutáneo que se asoció a un adenocarcinoma de pulmón.
Observación clínica
Varón de 78 años, ex fumador de más de 50 paquetes-año, diagnosticado 3 años antes de fibrosis pulmonar idiopática evolucionada, con insuficiencia respiratoria crónica. En una de las revisiones clínico-radiológicas se objetivó la aparición de una imagen seudonodular de 2,5 cm en el lóbulo superior izquierdo, que se asociaba a pequeño derrame pleural izquierdo, sin acompañarse de hemoptisis, síndrome constitucional ni síntomas localizados en otros órganos. En la exploración física se observaron unas lesiones pruriginosas y eritematovioláceas de carácter descamativo en las manos y los pies, así como hiperqueratosis y engrosamiento de la región periungueal de ambas extremidades, junto con eritema palmoplantar y en los pabellones auriculares (figs. 1 y 2). El paciente refería que habían aparecido unos 4 meses antes. No se registraron alteraciones analíticas clínicamente relevantes. Se realizó una fibrobroncoscopia, en la que se objetivó una lesión endobronquial con extensión submucosa en la entrada del lóbulo superior izquierdo, de la que se tomaron varias biopsias, cuyo resultado fue de adenocarcinoma.
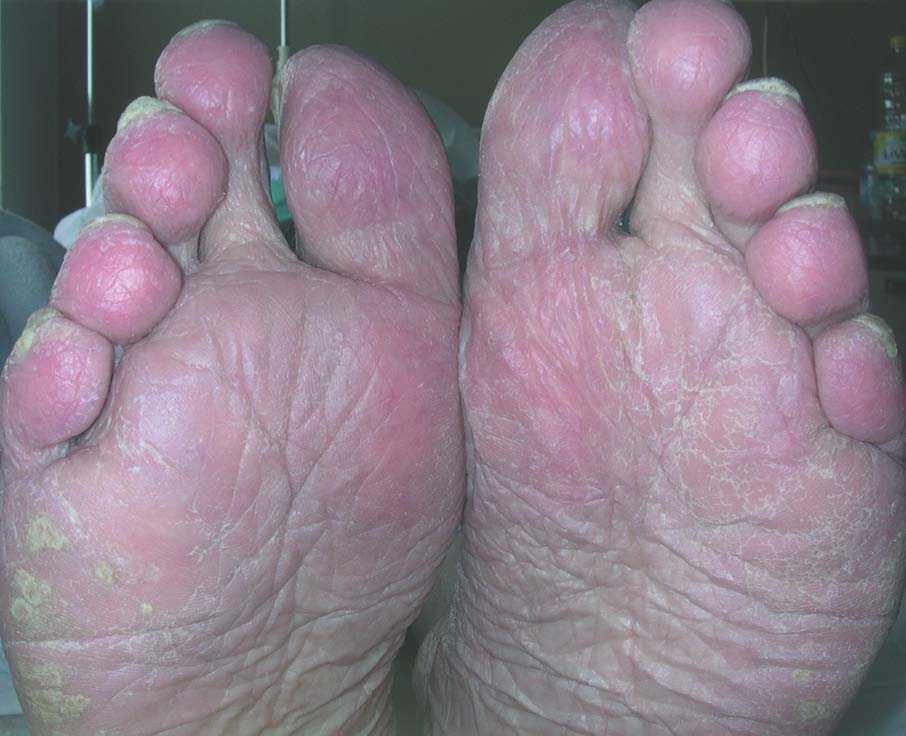
Fig. 1. Eritema y lesiones descamativas e hiperqueratósicas en la región plantar.
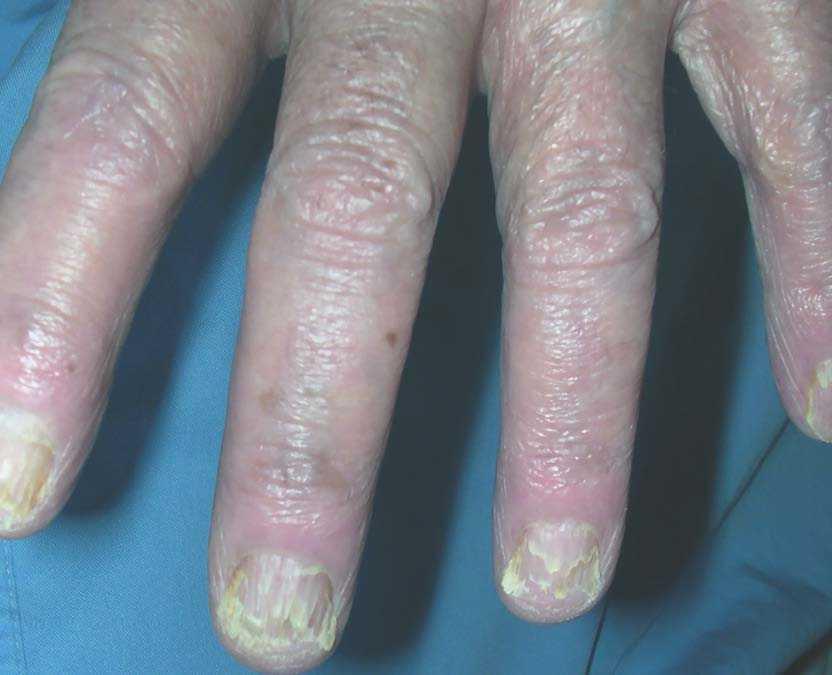
Fig. 2. Hiperqueratosis subungueal y onicólisis.
Discusión
Los carcinomas más frecuentemente asociados a la acroqueratosis paraneoplásica o síndrome de Bazex son los que afectan al tracto aereodigestivo superior5,6. En una revisión de la literatura médica realizada por Sarkar et al7, de 112 casos casi la mitad de las neoplasias se asentaba en la cavidad oral, faringe y laringe, y la mayoría correspondía a carcinomas epidermoides. Estos autores describieron 19 casos de cáncer de pulmón (sólo 3 de estirpe adenocarcinoma8) y 18 casos fueron carcinomas de origen desconocido con biopsia ganglionar positiva. Casos excepcionales se han descrito asociados a adenocarcinoma de próstata, gástrico, endometrio, epidermoide de vulva, hepatocarcinoma, carcinoide de pulmón9 y mama10. Desde el punto de vista clínico, los pacientes suelen ser varones, de raza blanca y con una edad media alrededor de 60 años. En el momento del diagnóstico presentan habitualmente afectación ganglionar metastásica, aunque las manifestaciones cutáneas pueden preceder varios meses a la manifestación propia del tumor.
Según las descripciones iniciales realizadas en 1965 por Bazex y Griffiths11, el síndrome comprende 3 fases. El primer estadio se caracteriza por una erupción simétrica y eritematodescamativa de carácter pruriginoso en las zonas distales de los dedos de las manos y de los pies, seguida de afectación de las orejas y la nariz. Posteriormente la zona periungueal se vuelve hiperqueratósica y las uñas, distróficas, con onicólisis asociada. En este estadio la enfermedad tumoral generalmente es asintomática y la clínica dermatológica suele preceder entre 2 y 6 meses a la misma. El segundo y tercer estadios corresponden a una afectación ganglionar con progresión de las lesiones cutáneas, que se extienden centrípetamente hacia el resto de las extremidades y el tronco. Desde el punto de vista histológico, las lesiones corresponden a hiper y paraqueratosis, acantosis y degeneración vacuolar de queratinocitos, con infiltración perivascular de predominio linfomonocitario en la dermis papilar5,10,12. La inmunofluorescencia directa puede mostrar depósitos focales de inmunoglobulinas, C3 o fibrina en la membrana basal5,6,10. En general, el diagnóstico es clínico, y se basa en la historia del paciente y en la característica distribución de las lesiones, por las que se pueden excluir otras enfermedades como la psoriasis acral, la pitiriasis rubra pilaris, el lupus eritematoso y la enfermedad familiar hiperqueratósica palmoplantar4,12-14.
La patogenia del síndrome es incierta. Varios autores señalan un mecanismo autoinmunitario, en el que habría un antígeno común entre las células tumorales y epidérmicas. Otras explicaciones se basan en la estimulación de factores de crecimiento (factor transformador del crecimiento tipo alfa)6,15 producidos por el tumor, que desencadenarían el efecto patogénico en la epidermis, fundamentalmente interviniendo en la generación de hiperqueratosis. Esta enfermedad cutánea no responde a tratamientos esteroideos ni queratolíticos, y las lesiones se resuelven en el 90% de los casos, parcial o totalmente, con el tratamiento efectivo de la neoplasia, aunque se han descrito casos de mejoría parcial con retinoides (etretinato)15. Las alteraciones ungueales son las que presentan una mejoría más lenta y pueden continuar alteradas tras los tratamientos del tumor16. En nuestro paciente, dadas la enfermedad de base (fibrosis pulmonar idiopática evolucionada) y la mala situación funcional, se optó por no recurrir a la quimioterapia o a la cirugía. El paciente falleció a los pocos meses por progresión de la enfermedad tumoral.
Correspondencia:
Dra. M.T. Río Ramírez.
Unidad de Neumología. Hospital Virgen de la Luz.
Hermandad de Donantes de Sangre, s/n. 16002 Cuenca. España.
Correo electrónico: mtrio@sescam.jccm.es
Recibido: 20-2-2006; aceptado para su publicación: 25-4-2006.







