









The objective of this analysis was the evaluation of a new national circuit used for diagnosing alpha1 antitrypsin deficiency (AATD) based on multiplex technology using online registration and mail posted samples from dried blood spots (DBS) and buccal swabs.
MethodsThis is an observational, ongoing study conducted in Spain since March 2018. Samples are coded on a web platform and sent by postal mail to the central laboratory. Allele-specific genotyping for the 14 most common mutations was done with the Luminex 200 Instrument System. Gene sequencing was done if none of the mutations were found and the AAT serum level was <60mg/dl, or by request from the clinician in charge.
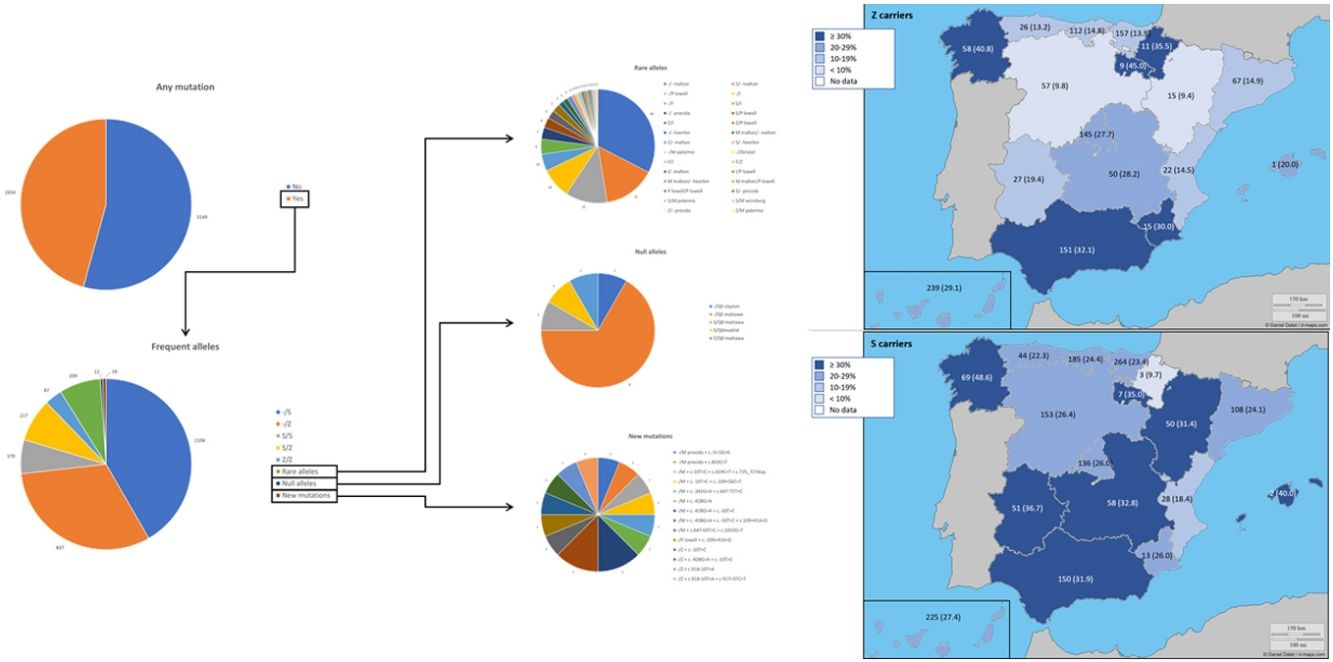
ResultsAt the time of the present report, 5803 (92.9%) samples were processed, 4984 (85.9%) from buccal swab and 819 (14.1%) from DBS. The prevalence of the frequent allele combinations were: MS 19.0%, MZ 14.4%, SS 2.9%, SZ 3.7%, and ZZ: 1.4%. Globally, Z carriers represented 20.0% and S carriers 26.6% of this population, with differences seen between regions. 209 (3.6%) were identified carrying rare alleles, 12 (0.2%) carrying null alleles and 14 (0.3%) new mutations were described. Respiratory diseases other than COPD, including poorly controlled asthma or bronchiectasis, also presented AATD mutations.
ConclusionsThe availability of a diagnostic system based on the simultaneous testing of 14 genetic variants from buccal swabs or DBS sent by postal mail and with web registration has proven to be useful, and the system can improve the timely diagnosis of AATD.
El objetivo de este análisis fue la evaluación de un nuevo circuito nacional utilizado para diagnosticar la deficiencia de alfa-1 antitripsina (DAAT) basado en tecnología multiplex con muestras de manchas de sangre seca (DBS, por sus siglas en inglés) y frotis bucales enviados por correo postal tras un registro previo en línea.
MétodosEste es un estudio observacional en curso que se está llevando a cabo en España desde marzo de 2018. Las muestras se codifican en una plataforma web y se envían por correo postal al laboratorio central. El genotipado de un alelo específico buscando las 14 mutaciones más comunes se realizó con el sistema Luminex® 200. Se realizó secuenciación génica si no se encontraba ninguna de las mutaciones y el nivel sérico de AAT era <60mg/dl, o por solicitud del médico responsable.
ResultadosEn el momento del presente informe se habían procesado 5.803 (92,9%) muestras, 4.984 (85,9%) de frotis bucal y 819 (14,1%) de DBS. La prevalencia de las combinaciones frecuentes de alelos fue: MS 19,0%, MZ 14,4%, SS 2,9%, SZ 3,7% y ZZ 1,4%. Globalmente, los portadores de Z representaron el 20,0% y los portadores de S el 26,6% de esta población, observándose diferencias entre las regiones. Se identificaron 209 (3,6%) portadores de alelos raros, 12 (0,2%) portadores de alelos nulos y se describieron 14 (0,3%) nuevas mutaciones. Otras enfermedades respiratorias que no eran EPOC, incluyendo el asma mal controlado o las bronquiectasias, también presentaron mutaciones DAAT.
ConclusionesLa disponibilidad de un sistema de diagnóstico con registro web basado en el análisis simultáneo de 14 variantes genéticas de frotis bucales o DBS enviados por correo postal ha demostrado ser útil, y el sistema puede mejorar el diagnóstico temprano de DAAT.
Despite the recommendations of the World Health Organization (WHO), major international scientific respiratory societies, and patient-focused associations regarding the active detection of alpha1 antitrypsin deficiency (AATD) in patients with chronic obstructive pulmonary disease (COPD),1–6 AATD continues to be an underdiagnosed condition. It is estimated that there are about 74,000 individuals with ZZ genotype in Western and Central European countries, of which 41% are identified, and that 44,000 live in North America, of which 24% are identified.7,8 Similarly, there are also about one and a half million individuals with SZ genotype, of which 48% live in Western European countries, 20% in the USA and 16% in South America.7,9 Given that AATD is associated with accelerated emphysema progression, and that delayed diagnosis is associated with worse COPD-related symptoms, functional status, and worsened air-flow obstruction,10 the importance of early detection of AATD in all adults with fixed air-flow obstruction and first-degree relatives of individuals with severe AATD is underscored.
Despite the variability of the different diagnostic algorithms,11,12 AATD diagnosis usually begins with the determination of AAT concentrations in serum samples or dried blood spots (DBS) followed by genotyping or phenotyping on the same sample. A recently reported multiplex technology which is able to simultaneously identify 14 mutations in the SERPINA1 gene has been used in Germany in DBS samples as a way to simplify and shorten the diagnostic workup of patients with suspected AATD.13 Additionally, the possibility of carrying out the genetic study using a sample taken by buccal swab has recently been described.14 Interestingly, the possibility of a complete diagnostic assessment of the deficiency from a buccal swab may facilitate the development of large-scale detection programs. Of note, although some initiatives are under evaluation in Italy,15 the performance of buccal swab samples in the diagnostic of AATD has not been reported in large populations.
In Spain, a new national circuit for diagnosing AATD was established in 2018. This new diagnostic circuit is coordinated by the Spanish Registry of patients with AATD (REDAAT) and is based on the genetic analysis of DBS or buccal swab sampling by a central laboratory (Progenika Biopharma, Derio, Vizcaya, Spain) using Luminex technology that identifies the 14 most frequent deficient variants simultaneously. The objective of this analysis was the evaluation of the new Progenika-REDAAT diagnostic circuit and to analyze the genotyping results of patients with suspected AATD throughout the country based on DBS and buccal swabs.
MethodsThis is an observational, ongoing study conducted in Spain, analyzing the anonymized data included in the Progenika web platform (https://grifolsalpha1test.com/) from March 23rd, 2018 to October 1st, 2019. The diagnostic kits for sampling with the DBS and buccal swab were provided free of charge by Grifols (Barcelona, Spain) upon request from the treating physicians. The samples were registered on the web platform through a unique code associated with each kit individually and sent by post to the reference laboratory at Progenika headquarters. To ensure confidentiality of the information, there was an intermediate company (Haiko Technologies, Bilbao, Spain) that acts as a firewall between Progenika and Grifols. The decision on whether to do a buccal or blood sample depended on the consensus decision between the responsible clinician and the patient. During the registration of the sample on the web, clinicians were asked to include some clinical data about the patient that included age, current smoking status (smoker, former smoker or never smoker), serum AAT level, and forced expiratory volume in 1 second (FEV1) expressed as a percentage of its predicted value. Clinicians were also asked to select the reasons for requesting the test from a list. This list included COPD, poorly controlled asthma, bronchiectasis, blood relatives of individuals with AATD, hepatopathy of unknown cause, shortness of breath and chronic cough in many family members, decreased protein alpha-1 peak in proteinogram, panniculitis or multiorgan vasculitis of unknown cause, and none of the above. The clinician could select more than one option from this list and was responsible for the accuracy of these diagnoses. These data were not compulsory to register the sample. Additionally, by identifying the user, the system recorded the hospital and province where the sample was registered. After receipt of the sample in the laboratory, there was an initial validation step to check that the sample arrived with good preservation status and the sample number was correctly identified on the sample and on the webpage.
Genetic testingAllele-specific genotyping was done with the Progenika A1AT Genotyping Test (PAGT). PAGT using OCR100 buccal swab has been cleared by FDA for prescription use and it is CE approved. The test allows simultaneous analysis of up to 384 samples per batch and is able to identify the 14 most frequent deficiency variants of the SERPINA1 gene, namely, PI*S, PI*Z, PI*I, PI*M procida, PI*M malton, PI*S iiyama, PI*Q0 granite falls, PI*Q0 west, PI*Q0 bellingham, PI*F, PI*P Lowell, PI*Q0 mattawa, PI*Q0 clayton, and PI*M heerlen. The test is CE marked (European Conformity) and United States Food and Drug Administration approved. The test is intended for use with genomic DNA extracted from human whole blood samples collected in K3-ethylenediaminetetraacetic acid tubes (EDTA), as DBS, or from human buccal swab samples.
The PAGT uses polymerase chain reaction amplification to obtain large amounts of the target sequences in the SERPINA1 gene. Extracted DNA is amplified and biotinylated by multiplex polymerase chain reaction and products are denatured and hybridized to oligonucleotide probes coupled to color-coded beads. Hybridized DNA is labeled with a fluorescent conjugate and the resulting signal is detected with a Luminex® 200 system. Raw data obtained are processed with the PAGT software in order to obtain the final report. This software algorithm converts the allelic variant genotypes into associated alleles. PAGT generates a simultaneous multiplex reaction in a single well, avoiding the need to run separate methods in parallel. When none of the 14 alleles studied was found the result was noted as negative and interpreted as an M allele, since the absence of any of these 14 alleles suggests with more than a 99% probability that the genotype corresponds to PI*M.
Gene sequencingThe Progenika clinical service laboratory proceeded to do SERPINA1 gene sequencing if they did not find any of the 14 mutations and the AAT serum level was <60mg/dl, or by request from the clinician in charge. The complete sequencing of the SERPINA1 gene (NM_001127701.1), including the 7 exons and flanking intronic zones, was performed by last generation sequencing-NGS (MiSeq Illumina). The obtained sequences were analyzed using the CLC Genomics Workbench (Qiagen) software to identify variants present in the sample. Only variants that could explain pathogenicity (variants validated in the literature or those that could affect protein activity or expression) were included in the report. Frequent polymorphisms or silent variants that do not affect splicing are considered to have no effect on protein levels or activity and are therefore not reported.
EthicsThe present study complies with the requirements of the Helsinki declaration for studies with human beings. The personal data of the patients were kept under strict confidentiality in compliance with the provisions of Organic Law 3/2018, of December 5, Protection of Personal Data and Guarantee of digital rights (LOPDGDD) and its development regulations, and in accordance with the provisions of Regulation (EU) 2016/679 of the European Parliament and of the Council of April 27, 2016 regarding the protection of natural persons with regard to the processing of personal data and the free circulation of these data. The biological samples related to the study were numbered with a code to guarantee the confidentiality of the sample and the associated clinical data. The relationship between this code and the medical record number was kept by the clinician under his sole responsibility and custody. Therefore, there was no data in the database that could be used to identify patients. The patients signed an informed written consent authorizing the clinicians to carry out the genetic study according to Spanish legislation.
StatisticsStatistical analyses were performed with IBS SPSS Statistics (IBM Corporation, Armonk, NY), version 26.0. Data were described using absolute counts with relative frequencies in parentheses for categorical variables. Quantitative data were summarized with the mean and the standard deviation (SD) in parentheses. Comparisons between categorical variables were done with the Chi-square test or Fisher exact test when applicable. Comparisons between quantitative variables between groups were done with unpaired Student's t-test, after checking the equality of the variances with the Levene's test. Alpha error was set at 5%.
ResultsDuring the study period, there were 6243 samples either recorded in the system or received in the laboratory, of which 5803 (92.9%) were processed at the time of the present report: 4984 (85.9%) from buccal swab and 819 (14.1%) from DBS (Fig. 1). One hundred seventy samples (2.7%) were not processed, with 7 (0.1%) of them due to the poor quality of the sample and 163 (2.6%) due to errors recording the identification code in the web. There was an uneven distribution of the samples throughout the country (Fig. 1S). Over time, there was a progressive increase in the inclusion of patients with a decrease during the summer months (online supplement Fig. 2S).

Subjects studied included 1424 (24.5%) current smokers, 2249 (38.8%) ex-smokers, and 2130 (36.7%) never smokers. Mean age was 54.7 (SD: 18.3) years. FEV1% predicted was available for 3229 (55.6%) cases with a mean value of 72.8 (SD: 24.3) %. AAT serum levels were available in 1637 (28.2%) cases, with a mean value of 83.6 (SD: 29.4) mg/dl (Fig. 2). The distribution of cases according to the different AAT cut-off values was: 207 (12.6%) were above 120mg/dl, 506 (30.9%) were between 90 and 120mg/dl, 693 (42.3%) were between 60 and 89mg/dl, and 231 (14.1%) were below 60mg/dl. Of the samples, 4564 (78.6%) were index cases and 1239 (21.4%) were relatives.

The reasons to include the patient in the diagnostic procedure according to the type of sample are summarized in Table 1. The most frequent reason was COPD in 2800 (48.2%) cases followed by blood relatives of individuals with AATD in 1239 (21.3%) cases. In 1073 (18.5%) cases, the clinicians reported more than one reason.
Reasons to include the patient in the screening procedure according to the type of sample.
| Dried blood spots (n=819) | Buccal swab (n=4990) | P value* | |
|---|---|---|---|
| Chronic Obstructive Pulmonary Disease (COPD) | 513 (62.6) | 2287 (45.9) | <0.001 |
| Poorly controlled asthma | 150 (18.3) | 606 (12.2) | <0.001 |
| Blood relatives of individuals with AATD | 66 (8.1) | 1173 (23.5) | <0.001 |
| Bronchiectasis | 20 (2.4) | 193 (3.9) | 0.044 |
| Hepatopathy of unknown cause | 10 (1.2) | 47 (0.9) | 0.455 |
| Shortness of breath and chronic cough in many family members | 8 (1.0) | 76 (1.5) | 0.224 |
| Decrease in alpha-1 peak in proteinogram | 7 (0.9) | 218 (4.4) | <0.001 |
| Panniculitis or multiorgan vasculitis of unknown cause | 0 (0) | 4 (0.1) | 0.417 |
| None of the above | 0 (0) | 0 (0) | – |
| Not registered | 109 (13.3) | 655 (13.1) | 0.896 |
Data expressed as absolute (relative) frequencies. Percentages referred to the total number per sample type.
The average number of days of the different procedural steps is shown in the online supplement Fig. 3S. While the samples were genotyped when received at the laboratory, for sequencing it was expected to have a minimum number of samples to be processed together. Consequently, sequencing times were longer. Gene sequencing was done in 77 (1.3%) cases (online supplement Table 1S). In all the cases the sequencing results were consistent with the A1AT Genotyping Test results. In 22 (28.6%) cases, sequencing revealed an additional mutation from direct genotyping.
The complete list and distribution of mutations is shown in Fig. 3. There were 3150 (54.3%) samples carrying no mutations, 2417 samples (41.6%), carrying frequent mutations (S or Z), 209 samples (3.6%) carrying rare alleles, 12 samples (0.2%) carrying a null allele, and 16 samples (0.3%) carrying 14 new mutations. The description of these 14 new mutations is provided in Table 2. The complete list of mutations found by AAT serum levels is shown in the online supplement Table 2S. The prevalence of the frequent allele combinations in this selected population was: MS 19.0%, MZ 14.4%, SS 2.9%, SZ 3.7%, and ZZ: 1.4%. Considered globally, 20.0% of the individuals were Z carriers and 26.6% of the individuals were S carriers. There were 5 cases with AAT levels<60mg/dl and no mutations by direct genotyping, in which sequencing confirmed the absence of any mutations. The distribution of Z and S carriers in the different regions of the country is shown in Fig. 4 (p<0.001 between regions and provinces). The distribution of the mutation groups according to the reason for genotyping is shown in Table 3. Individuals with respiratory diseases other than COPD, including poorly controlled asthma or bronchiectasis, also presented with AATD mutations. Index cases presented with a different distribution of clinical variables and mutations as compared to relatives (Table 4).

Description of the new mutations found.
| Nucleotide change | Genome position (GRCh38) | Aminoacid change in the mature protein | Mutation type | Predicted protein activity | Pathogenicity | SNP code | ClinVar code | Exon (NM_001127701.1) |
|---|---|---|---|---|---|---|---|---|
| c.918-10T>A | g.94379621 | NA | Splicing | Probably reduced | Probably pathogenic | ND | Not Reported | intron5 |
| c.917+37C>T | g.94380834 | NA | Nucleotide change | Probably normal | Probably not pathogenic | ND | Not Reported | intron5 |
| c.-428G>A | g.94390547 | NA | 5 Prime UTR | Unknown | Unknown | rs56367058 | Not Reported | exon1 |
| c.-10T>C | g.94388565 | NA | 5 prime UTR | Unknown | Unknown | rs11558258 | 315034 | exon3 |
| c.-109+41A>G | g.94388778 | NA | Nucleotide change | Probably normal | Probably not pathogenic | rs55967149 | 315042 | intron2 |
| c.647-69T>C | g.94381210 | NA | Nucleotide change | Probably normal | Probably not pathogenic | rs55695365 | Not Reported | intron4 |
| c.1033G>T | g.94379496 | Val321Phe | Aminoacid change | Unknown | Unknown | rs761711628 | Not Reported | exon6 |
| c.-342G>A | g.94390461 | NA | 5 prime UTR | Unknown | Unknown | rs78070723 | Not Reported | exon1 |
| c.647-71T>C | g.94381212 | NA | Nucleotide change | Probably normal | Probably not pathogenic | rs77012599 | Not Reported | intron4 |
| c.-109+56C>T | g.94388763 | NA | Nucleotide change | Probably normal | Probably not pathogenic | rs55973910 | 315041 | intron2 |
| c.424C>T | g.94382814 | Leu118Leu | Nucleotide change | Probably normal | Probably not pathogenic | rs20546 | 178806 | exon4 |
| c.725_727dup | g.94381061_94381063 | Val218_Pro219insLeu | In-frame indel | Probably reduced | Probably pathogenic | ND | Not Reported | exon 5 |
| c.853C>T | g.94380935 | Gln261* | Nonsense | No activity | Pathogenic | rs1480528329 | Not Reported | exon5 |
| c.-5+1G>A | g.94388559 | NA | Splicing | Probably reduced | Probably pathogenic | rs775786225 | Not Reported | intron3 |
NA: not available; ND: not described.
 and the number of maps used is limited to 10 per publication.")
Distribution of Z and S carriers in the country. Results expressed as absolute count with percentages of the samples in the region in parentheses. The map used to make this figure is protected by copyright. They are modifiable and free for any use as long as the exact URL where the original map comes from is mentioned (https://d-maps.com/) and the number of maps used is limited to 10 per publication.
Distribution of mutation groups according to the reason for genotyping.
| No mutations | Frequent mutations | Rare alleles | Null alleles | New mutations | |
|---|---|---|---|---|---|
| Chronic Obstructive Pulmonary Disease (COPD) (n=2800) | 1817 (64.9) | 907 (32.4) | 68 (2.4) | 3 (0.1) | 5 (0.2) |
| Poorly controlled asthma (n=756) | 492 (65.1) | 248 (32.8) | 16 (2.1) | 0 (0) | 0 (0) |
| Bronchiectasis (n=213) | 105 (49.3) | 103 (48.4) | 4 (1.9) | 0 (0) | 1 (0.5) |
| Blood relatives of individuals with AATD (n=1239) | 363 (29.3) | 789 (63.7) | 80 (6.5) | 2 (0.2) | 5 (0.4) |
| Hepatopathy of unknown cause (n=57) | 16 (28.1) | 34 (59.6) | 5 (8.8) | 1 (1.8) | 1 (1.8) |
| Shortness of breath and chronic cough in many family members (n=84) | 40 (47.6) | 41 (48.8) | 2 (2.4) | 0 (0) | 1 (1.2) |
| Decrease in alpha-1 peak in proteinogram (n=225) | 45 (20.0) | 158 (70.2) | 17 (7.6) | 3 (1.3) | 2 (0.9) |
| Panniculitis or multiorgan vasculitis of unknown cause (n=4) | 2 (50.0) | 1 (25.0) | 0 (0) | 1 (25.0) | 0 (0) |
| Not registered (n=764) | 417 (54.6) | 308 (40.3) | 31 (4.1) | 5 (0.7) | 3 (0.4) |
Data expressed as absolute numbers with percentages in parenthesis referred to the total number per row. AATD=alpha 1 antitrypsin deficiency.
Differences between index cases and relatives of individuals with alpha 1 antitrypsin deficiency.
| Index cases (n=4564) | Relatives (n=1239) | P value* | |
|---|---|---|---|
| Buccal swab | 3811 (83.5) | 1173 (94.7) | <0.001 |
| Current smokers | 1205 (26.4) | 219 (17.7) | <0.001 |
| Age (years) | 59.4 (14.7) | 37.4 (19.9) | <0.001 |
| Primary care | 93 (2.0) | 9 (0.7) | 0.002 |
| AAT available | 1342 (29.4) | 295 (23.8) | <0.001 |
| AAT (mg/dl) | 85.6 (29.4) | 77.9 (28.6) | <0.001 |
| FEV1 available | 2944 (64.5) | 285 (23.0) | <0.001 |
| FEV1 (%) | 70.0 (23.7) | 94.8 (18.3) | <0.001 |
| Cases with sequencing done | 65 (1.4) | 10 (0.8) | 0.088 |
| Cases with no mutation found | 2786 (61.0) | 363 (29.3) | <0.001 |
| Cases with frequent mutations | 1641 (36.0) | 791 (63.8) | <0.001 |
| Cases with rare alleles | 116 (2.5) | 78 (6.3) | <0.001 |
| Cases with null alleles | 10 (0.2) | 2 (0.2) | 0.692 |
| Cases with new mutations | 11 (0.2) | 5 (0.4) | 0.333 |
Data expressed as absolute numbers with percentages in parenthesis referred to the total number per column or mean with standard deviation in parentheses depending on the nature of the variable. AAT=alpha 1 antitrypsin; FEV1=forced expiratory volume in 1 second.
This report summarizes the evaluation of the new Progenika-REDAAT diagnostic circuit in Spain used for patients with suspected AATD. The circuit is based on a diagnostic system that allows the simultaneous testing of 14 genetic variants using either DBS or buccal swabs utilizing online registration and mail posted samples. Our results show that the new diagnostic circuit is feasible, with the majority of samples arriving at the reference laboratory in good condition to allow for genotyping. Additionally, we report the results of the buccal swab sample genotyping in a real-world setting from a large population sample.
The availability of a new diagnostic circuit with easy accessibility, without added cost and with all the ethical guarantees of confidentiality is an important pillar for improving the diagnosis of AATD. The main strengths of the circuit are the national coverage and the capacity of the laboratory to genotype and analyze all samples in a short period of time, with the possibility of gene sequencing under demand or after unexpected results from both index cases or relatives. Another strength of the circuit is the possibility of carrying out the diagnostic study from any medical clinic, without the need to refer the patient to a center of greater complexity. In this regard, buccal swabs facilitate the collection of samples in the clinical practice. Additionally, prior to the implantation of the diagnostic circuit, validation studies were carried out at Progenika to collect samples for genotyping with the A1AT Genotyping test. The validity of the results obtained has been demonstrated by comparing with Sanger sequencing as a standard Gold technique. Further, this system has been successfully used in other European countries.13,16 This circuit, therefore, could favor the early diagnosis of the disease, since many patients with mild COPD are often managed in the outpatient setting. The main limitation of the circuit that we have found is the registration of the samples on the website. Despite the potential advantage of having an online system for data registration, this system depends on the clinician correctly registering the sample code on the website to relate the results to the clinical data. In addition, the clinician must record the connection of the sample code and the patient's clinical record number. To facilitate this work, the web platform has a series of documents available to the clinician, including informed consent and a spreadsheet to keep track of registered patients and their correlation with the clinical record numbers. A note of caution should be considered for the cases with hepatopathy of unknown cause. The clinicians participating in this circuit were mostly pulmonologists or general practitioners. Therefore, cases with hepatopathy of unknown cause may be under-represented. The incorporation of liver disease specialists to the circuit would contribute to the detection of cases of AATD in this clinical context. Each individual clinician should put in a balance the above commented strengths and limitation and decide about the best diagnostic procedure at each site. In any case, we believe that this initiative will rise interest in the disease.
Interestingly, relatives presented a higher proportion of mutations, as expected for being a highly selected population. Additionally, this could possibly be related to a very active behavior in some regions or the inclusion of a less invasive, faster and simpler technique that could have induced an easier study for relatives.
The distribution of the mutations found in our program is somewhat different from that described in other European detection programs. In those initiatives, samples with identified mutations varied 87.6% in Poland, 62.9% in Germany or from 72.8 to 85.1% in Ireland.17–19 The distribution of S and Z alleles in Spain are in line with European reported prevalence of S and Z alleles where there is an increase of the S allele in Spain and Portugal as compared to other European countries.7,20–22 In Spain, a previous analysis identified the greatest frequency for S allele type is in the North-West of the country, while the greatest frequency for Z allele type has been reported in newborn infants from Valladolid (Center-West of Spain) and in a randomly selected general population of Asturias (North of Spain). Our current analysis updates the distribution of S and Z alleles in the Spanish population.20,21,23 A previous review article on the prevalence of AATD alleles in Spain23 estimated that there could be more than 9 million individuals carrying these alleles, of which 80% would be the PIMS phenotype and the rest would be PIMZ (13%), PISS (4.7%), PISZ (1.6%) and PIZZ (0.1%). We can now give real figures on the prevalence of theses alleles in the explored population with some changes from estimated at those years.AATD was detected in conditions other than COPD. In Spain, a recent cross-sectional cohort study evaluated 648 patients with house dust mite allergic asthma with different degrees of severity in the Canary Islands. The authors found 145 asthmatic patients (22.4%) with at least one mutated allele (S or Z), but neither association between deficient AAT genotypes or serum AAT and development of severe asthma, or correlation between AAT levels and FEV1 was observed.24 This study confirms previous observations that failed to identify higher prevalence of deficient phenotypes in adults with asthma or any influence of phenotypes on the severity of the disease.25 Further, AAT heterozygosis does not seem to be an important risk factor of persistent airflow limitation in patients with asthma.26 On the other hand, some AAT polymorphisms have been specifically described in asthma27,28 and some authors suggest that specialists in Allergy and Immunology should consider and screen for AATD in patients with asthma in whom spirometry does not return to normal.29 In our cohort, we have been able to identify severe mutations in patients with poorly controlled asthma, suggesting that this population should also be screened. Another respiratory comorbidity explored in our analysis was bronchiectasis. In addition to the existence of mutations described in this group,30,31 bronchiectasis patients with AATD seem to have some unique characteristics. Specifically, it has been described that mycobacterial infection is more frequent in these patients.32 An increased neutrophil expression of elastase mRNA has been described as a potential explanation for the early-age onset of bronchiectasis in AATD.33
Although the lower limit of normal AAT by nephelometry is 90mg/dl, our results indicate that severe deficient mutations can also be detected above this level. The variability of AAT levels for the different AATD genotypes has been described34 and may also be influenced by increased systemic inflammation.35 Consequently, the use of a higher than normal cut-off AAT value could be explored as a threshold value to study the possible presence of a deficient allele. Interestingly, we have been able to detect mutations even with AAT levels above 120mg/dl. In these cases, however, we cannot be sure that these AAT values are not high as a result of some systemic inflammatory reaction, since we did not have a C-reactive protein determination or another acute phase reactant available.36
One note to consider is that the multiplex system studies the 14 most common mutations. Therefore, the identification of the Pi*M is done by exclusion, since the absence of any of these 14 alleles suggests with more than a 99% probability that the genotype corresponds to PI*M. However, in case of a discrepancy with AATD serum levels, or by request from the clinician, the gene could be sequenced. Interestingly, this scenario only happened in a small number of cases during our study, highlighting that the 14 genotypes studied include more than 99% of deficient variants observed in the world.
In conclusion, our results report on a new diagnostic circuit capable of conducting simultaneous testing of 14 genetic variants from DBS and buccal swabs for diagnosing AATD. While additional enhancements could improve this new tool, the availability of a diagnostic system which evaluates buccal samples or DBS sent by postal mail with web registration has proven to be adequate and can improve the timely diagnosis of AATD. This system could also be exported to other countries where there is a need for a better and faster way to diagnosis AATD.
FundingThis project is entirely funded by Grifols.
Authors’ contributionsJLLC performed the statistical analysis, included cases and wrote the manuscript; FCM, MTD, AMG, JLGR, IC, MC and MM included cases, revised the draft and contributed with the writing; LO and NR performed the laboratory analysis, revised the draft and contributed with the writing.
Conflicts of interestJLLC has received honoraria during the last three years for lecturing, scientific advice, participation in clinical studies or writing for publications for (alphabetical order): AstraZeneca, Boehringer Ingelheim, Chiesi, CSL Behring, Laboratorios Esteve, Laboratorios Ferrer, Gebro Pharma, GlaxoSmithKline, Grifols, Menarini, Novartis, Rovi, and Teva. FCM has received speaker fees from AstraZeneca, Bial, Boehringer Ingelheim, Chiesi, CSL Behring, Laboratorios Ferrer, Gebro Pharma, GlaxoSmithKline, Grifols, Laboratorios Esteve, Menarini, Mundipharma, Novartis, Praxis, Rovi, Sandoz, TEVA, Vertex and Zambon. MTD has received honoraria from Chiesi, CSL Behring, Laboratorios Esteve, Laboratorios Ferrer, GlaxoSmithkline and Grifols. MC has received speaker fees from Boehringer Ingelheim, AstraZeneca, GlaxoSmithKline, Menarini, and Grifols and consulting fees from GlaxoSmithKline, Gebro Pharma and Novartis. MM has received speaker fees from AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, Menarini, Rovi, Bial, Sandoz, Zambon, CSL Behring, Grifols and Novartis, consulting fees from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Bial, Gebro Pharma, CSL Behring, Laboratorios Esteve, Laboratorios Ferrer, Mereo Biopharma, Verona Pharma, Teva, pH Pharma, Novartis and Grifols and research grants from GlaxoSmithKline and Grifols.
The authors want to show their appreciation to all the clinicians responsible for the collection of samples and their determined effort to improve the diagnosis of patients with AATD. The authors also want to show their appreciation to MaryJane Silvey for having revised and improved the English of the manuscript.







