

Primary immunodeficiencies (PIDs) are a heterogeneous group of more than 400 inherited immune system disorders, with overall prevalence 1/1000–1/5000.1 At any age, recurrent-to-persistent respiratory infections are often the first presenting sign of PIDs.2 Poor defense from opportunistic or non-opportunistic pathogens, as well as non-infectious complications may significantly impact morbidity and mortality of the conditions, even when early detected.3
In PIDs, type, outcome and severity of the underlying defect might influence type and severity of patients’ respiratory phenotypes, but only few studies documented this.4
In this report, we compared the respiratory manifestations and the chest imaging findings from a cohort of pediatric and adult patients from a tertiary level hospital, a major referral for PID, located in Campania region, in Southern Italy. In order to describe the respiratory phenotypes in different PID groups and to investigate their prevalence, we conducted a retrospective study over a three-year period, from mid-2018 to mid-2020 and created a database of 269 patients with PID including 182 children (mean age, 9±4 years; 67% of the total) and 87 adults (mean age, 20±6.5 years; 33% of the total). According to the underlying diagnosis, patients were allocated to three groups: cellular immunity defects [Group 1, n=48, 17.9% of the total, including Ataxia–Teleangiectasia (A–T), partial DiGeorge syndrome (pDGS), or Severe Combined Immune-deficiencies (SCID) before treatment]; humoral immunity defects [Group 2, n=203, 75.5% of the total, including Common Variable Immune-deficiency (CVID), X-linked Agammaglobulinemia (XLA), or selective IgA Deficiency (sIgAD)]; innate immunity defects [Group 3, n=18, 6.6% of the total, including Chronic Granulomatous Disease (CGD), STAT1 gain of function, hyper IgE Syndrome, MYD88 Deficiency, or congenital neutropenia]. We analyzed variables including gender, type of PID, age at diagnosis, age at onset of respiratory symptoms, diagnostic delay (the time elapsed between the onset of respiratory symptoms and the diagnosis of PID), history of upper (i.e. rhinosinusitis and/or otitis) or lower (i.e. bronchitis and/or pneumonia) airway infections, chest imaging phenotypes (at X-ray or Computed Tomography or Magnetic Resonance Imaging).5 In our study population, Groups 1 and 3 included only children, while Group 2 was composed of both pediatric and adult patients with humoral immunity defects.
Comparisons between groups were performed applying t-test for numerical variables and chi-test for categorical variables.
The age at onset of respiratory symptoms was significantly higher in Group 2 than Groups 1 and 3 (P<.001; Fig. 1A). In more than half of the cases, a diagnosis of cellular immunity defect was established, in nearly 90%, of cases in the first decade (Fig. 1B). A wide variability of age at diagnosis was found in cases with humoral immunity defects, while patients with defects of innate immunity were diagnosed always before adolescence. The diagnostic delay was lower in patients with cellular immunity defects than in other groups (P<.01; Fig. 1C). Rhinosinusitis was more common in Group 2, with a significant difference when compared to Groups 1 and 3 (P<.05), and otitis was more frequent in Group 1. No significant difference in the prevalence of lower airway infections was found among all groups (Fig. 1D).
 Age of onset of respiratory symptoms. (b) Age distribution at diagnosis according to different IEI. (c) Diagnostic delay. (d) Distribution of respiratory infections among the three groups. (e) Upper respiratory tract infections within Group 1. (f) Lower respiratory tract infections within Group 1. (g) Prevalence of rhinosinusitis in Group 2. (h) Distribution of respiratory infections within Group 3. *P-value≤.05, ** P-value≤.01, *** P-value≤.0001.")
(a) Age of onset of respiratory symptoms. (b) Age distribution at diagnosis according to different IEI. (c) Diagnostic delay. (d) Distribution of respiratory infections among the three groups. (e) Upper respiratory tract infections within Group 1. (f) Lower respiratory tract infections within Group 1. (g) Prevalence of rhinosinusitis in Group 2. (h) Distribution of respiratory infections within Group 3. *P-value≤.05, ** P-value≤.01, *** P-value≤.0001.
In order to investigate whether a different localization of airway infections in each disease group was associated with a specific immunological defect, we compared the prevalence within subgroups. In Group 1, upper airway infections were more frequent in A-T patients, while lower airway infections were more prevalent among SCID patients (Fig. 1E–F). Within the pediatric patients from Group 2, we compared the prevalence among XLA, CVID and sIgAD, and found that rhinosinusitis was more prevalent in the CVID subgroup (P<.05) (Fig. 1G). Within Group 3, patients with CGD or other innate immunity disorders showed higher prevalence of pneumonia in the CGD subgroup (Fig. 1H).
Of 269 patients, 197 (72.3%) underwent chest imaging examinations at the time of database creation. Four major categories of radiological changes were analyzed: consolidation, bronchiectasis, interstitial lung disease and Granulomatous and Lymphocytic Interstitial Lung Disease (GLILD).6
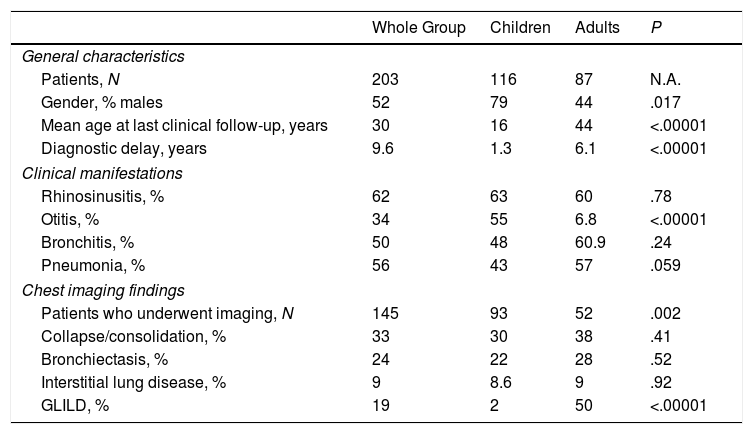
Consolidation was more common among patients with cellular and innate immunity defects than in those with humoral immunity defects (P<.05). Prevalence of bronchiectasis was significantly higher in patients with humoral immunity defects compared to cellular immunity defects (P<.05). As the only group including both pediatric and adult patients is Group 2, we compared the general characteristics, clinical manifestations and chest imaging findings between the two age subgroups (Table 1). As expected, adults with humoral immunity defects had a straightforward delay in the diagnosis of PID (6.1 versus 1.3 years in the pediatric population; P<.00001), while children showed a fairly higher rate of otitis than adults (55% versus 6.8%, respectively; P<.00001). Finally, no noteworthy difference was found in the prevalence of radiological findings between the two age subgroups, except for GLILD that was found in 28 CVID patients of which 93% were adults (Table 1).
General Characteristics, Clinical Manifestations and Chest Imaging Findings of Group 2 Including Pediatric and Adult Patients With Humoral Immunity Defects; the Comparison Between the Two Age Groups, Calculated Using Either t-test or chi-test, Are Shown on the Right Column.
| Whole Group | Children | Adults | P | |
|---|---|---|---|---|
| General characteristics | ||||
| Patients, N | 203 | 116 | 87 | N.A. |
| Gender, % males | 52 | 79 | 44 | .017 |
| Mean age at last clinical follow-up, years | 30 | 16 | 44 | <.00001 |
| Diagnostic delay, years | 9.6 | 1.3 | 6.1 | <.00001 |
| Clinical manifestations | ||||
| Rhinosinusitis, % | 62 | 63 | 60 | .78 |
| Otitis, % | 34 | 55 | 6.8 | <.00001 |
| Bronchitis, % | 50 | 48 | 60.9 | .24 |
| Pneumonia, % | 56 | 43 | 57 | .059 |
| Chest imaging findings | ||||
| Patients who underwent imaging, N | 145 | 93 | 52 | .002 |
| Collapse/consolidation, % | 33 | 30 | 38 | .41 |
| Bronchiectasis, % | 24 | 22 | 28 | .52 |
| Interstitial lung disease, % | 9 | 8.6 | 9 | .92 |
| GLILD, % | 19 | 2 | 50 | <.00001 |
Abbreviations: GLILD, granulomatous lymphocytic interstitial lung disease.
Overall, our report reveals, as expected, that in cases with the most severe PID, such as cellular or innate immunity defects, respiratory symptoms start early.7 This resulted in a prompt immunological diagnosis. Patients with humoral immunity defects usually present with mild upper respiratory tract infections which might be, indeed, overlooked, thus delaying the referral to the immunology center. The association with other non-immunological features may also reduce the diagnostic delay in selected cases (A-T or DGS). Moreover, the finding that rhinosinusitis is the most prevalent respiratory manifestation in CVID (Group 2) or A-T patients (Group 1) might be explained by the long-lasting T-cell defect in both disorders, rather than by the severity of T-cell impairment or the selective humoral immunity defects. While we did not find any significant difference in lower respiratory tract infections distribution among all groups, rhinosinusitis associated with GLILD appeared a distinctive feature of the respiratory phenotype of CVID. Early recurrent sinopulmonary infections are extremely common in CVID, and a high proportion of cases may develop chronic lung disease with bronchiectasis.3 A host of other systemic manifestations, including autoimmune disorders and lymphoproliferation may also occur which significantly worsen patients’ morbidity and mortality. Prognosis of CVID is largely affected by GLILD, a lymphoproliferative disorder characterized by progressive lung damage including airway disease and the need for continuous chest treatment.8,9
Although the hallmarks of respiratory manifestations in the different groups of PID are already known, our report, through a stratified sampling method on a single-center cohort including both pediatric and adult patients, highlights that rhinosinusitis and GLILD are almost exclusively found in CVID, suggesting that the contextual presence of a cell-mediated defect of long duration in association with the humoral defect is necessary for its development. Hopefully, the precocious identification of the lung disease may help improve the final outcome of the disease and more importantly may significantly impact the quality of life of patients with PID.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflicts of InterestThe authors declare to have no conflict of interest directly or indirectly related to the manuscript contents.







