





Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible and frequently fatal disease. Currently there are national and multinational registries in Europe, United States, Australia and China to better understand the magnitude of the problem and the characteristics of the IPF patients. However, there are no national or regional registries in Latin America, so the objective of this study was to carry out a Latin American registry that would allow the identification of IPF patients in our region.
MethodologyA system consisting of 3 levels of control was designed, ensuring that patients met the diagnostic criteria for IPF according to international guidelines ATS/ERS/ALAT/JRS 2011. Demographic, clinical, serological, functional, tomographic, histological and treatment variables were recorded through a digital platform.
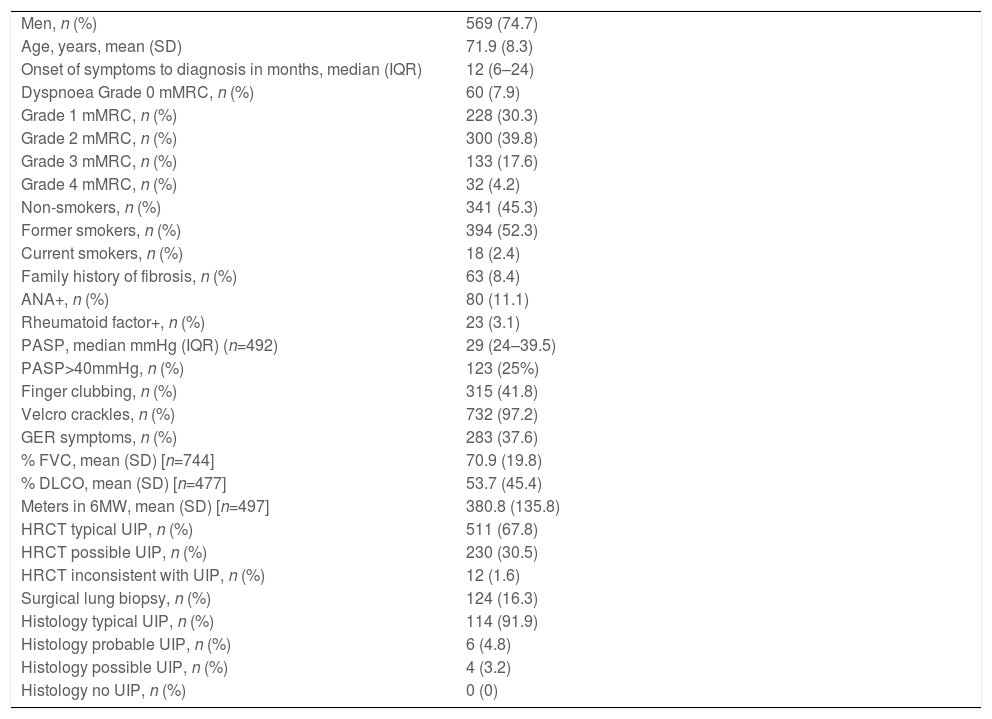
Results761 IPF patients from 14 Latin American countries were included for analysis, 74.7% were male, with a mean age of 71.9+8.3 years. In general there was a long period of symptoms before definitive diagnosis (median 1 year). In functional tests, an average reduction of FVC (70.9%) and DLCO (53.7%) was detected. 72% received at least one antifibrotic drug (pirfenidone or nintedanib) and 11.2% of the patients had an acute exacerbation, of which 38 (45.2%) died from this cause.
ConclusionsLike other registries, we found that there is difficulty in the recognition and excessive delay in the diagnosis of IPF in Latin America. Most of the patients in REFIPI received antifibrotics; these were well tolerated and associated with fewer adverse events than those reported in clinical trials.
Idiopathic pulmonary fibrosis (IPF) is defined as a specific form of chronic, progressive fibrosing interstitial pneumonia of unknown cause. It is usually irreversible, progressive, and fatal within a few years.1,2
IPF presents with different clinical phenotypes, making it particularly difficult to study, even in specialized centres.3 Because of this, national and even regional registries have been developed to gain a clearer understanding of the clinical, radiological, functional and morphological characteristics of IPF.
National registries, each with its own methodology, have now been created in Germany, Sweden, the United Kingdom, Greece, Spain, Australia, India and China.4–12 Multinational registries have only been developed in Europe.10,13
One of the first IPF registries to be created was the Australian Registry, which was compiled between 2012 and 2014 and included 359 participants.7 In the first description of the German registry,4 published in 2014, the authors reported that 34.1% of their 502 incident and prevalent patients required surgical lung biopsy. They also observed that even in a developed country such as Germany, only 2.8% of the study cohort was actually on a lung transplant list, although 58.6% of the patients were potential transplant candidates based on their age (less than 65 years) and disease severity (FVC<50% and/or DLCO<40%).
In 2014 Ryerson et al. called for the immediate establishment of a global IPF registry.14 Aware of the pressing need for new registries, we set out to establish the first Latin American IPF registry, the REFIPI (Latin American idiopathic pulmonary fibrosis registry). This registry was created at the initiative of the Department of Interstitial Lung Diseases (ILD) of the Latin American Thoracic Association (ALAT) for the purpose of studying the Latin American population of patients with IPF, supplementing the results of real-world evidence trials on the efficacy and safety of antifibrotic treatments, and comparing our results with other international registries to help create a “global IPF registry”.
The main objective of the REFIPI was to determine the demographic, clinical, functional and radiological characteristics of patients with IPF in Latin America at the time of diagnosis and over the course of their disease, and to record the treatments used, their efficacy and their safety in clinical practice.
MethodsStudy designA central executive committee made up of former directors and the deputy director and director of the ILD section of ALAT was initially formed in 2017 to define the variables to be studied in the REFIPI. A virtual REFIPI platform was then created for uploading patient data (https://alatorax.org/en/login).
An invitation was posted on the ALAT website and also sent by email to all pulmonologists associated with ALAT's ILD section inviting them to participate freely and voluntarily in the project.
National coordinators were put in charge of promoting the registry in their country and encouraging pulmonologists to upload data from their patients.
A 3-tier verification system was designed to ensure that patient data uploaded on the registry met the diagnostic criteria for IPF according to ATS/ERS/ALAT/JRS international guidelines14 (Fig. 1).
, description of each tier, and reasons for excluding cases at each tier level.")
1st tier: Treating physician, responsible for entering patient data.
2nd tier: Country coordinator.
3rd tier: ALAT Executive Committee on Diffuse Interstitial Lung Diseases.
From 1 November 2017 to November 2019, patients over 50 years of age diagnosed with IPF according to ATS/ERS/ALAT/JRS 2011 criteria14 were included, regardless of their disease progression.
Patients who had not given their informed consent, cases that lacked the basic variables for a diagnosis of IPF, and/or were found to have another ILD diagnosis after second and/or third tier verification were excluded from the registry.
All participating physicians complied with their national regulations, and each centre was asked to obtain approval from their ethics committee and the informed consent of each patient submitted to the registry, in accordance with the Declaration of Helsinki principles of good clinical practice.
Study variablesDemographic, clinical, blood, lung function, CT scan, histology, and treatment variables were collected:
Blood: Rheumatoid factor (RF) was recorded as positive/negative according to the reference values used in each method. Samples were tested for antinuclear antibodies (ANA) using indirect immunofluorescence (IIF), and positivity was defined as a titre equal to or greater than 1:80.
CT scan: High-resolution computed tomography (HRCT) patterns were categorized as typical usual interstitial pneumonia (UIP), possible UIP, and inconsistent with UIP, following the recommendations of the ATS/ERS/JRS/ALAT 2011 guidelines.1
Lung function: Forced vital capacity (FVC % predicted), diffusing capacity for carbon monoxide (DLCO % predicted) adjusted for haemoglobin, and distance achieved in the six-minute walk (6MW) test. All these variables were mandatory at the time of diagnosis but voluntary during follow-up, with the frequency of testing being decided by each pulmonologist.
Histology: The histology pattern was categorized according to the ATS/ERS/JRS/ALAT 2011 guidelines1 as typical usual interstitial pneumonia (UIP), probable UIP, possible UIP, and inconsistent with UIP.
Treatment: Variables related to treatment for gastro-oesophageal reflux (GER), use of corticosteroids and/or immunosuppressants, and treatment with pirfenidone and/or nintedanib were recorded. Tolerance of antifibrotics was evaluated on the basis of the different antifibrotic-related adverse events recorded from the date the drug was started until the last visit, death or suspension of the drug. Significant elevation of liver enzymes was defined as ≥3 times the normal value in patients who reported symptoms, or ≥5 times in those without symptoms.
Disease progression: Date and cause of death or lung transplantation and acute exacerbation according to recent criteria were recorded.15 Follow-up was defined as the time elapsed from the date of diagnosis to the date of the last consultation or the date of death/transplantation, as appropriate.
Statistical analysisFor the descriptive analysis, quantitative variables were reported as mean±standard deviation or median with interquartile ranges 25–75, depending on the distribution of the sample. The distribution of the variables was analysed using histograms and skewness and kurtosis values. Categorical variables were described as absolute and relative frequencies (proportion). Qualitative variables were analysed using Fisher's exact test or the Xi squared test, as appropriate. Logistic regression models were used to adjust for possible confounders. The odds ratio (OR) and confidence interval (95% CI) were reported for the variables used in the final models. Goodness of fit was evaluated using the Hosmer–Lemeshow test. Finally, all statistical calculations were performed using STATA 13.0 (Stata Corporation, College Station, TX, USA).
ResultsOne hundred and forty six doctors from 14 Latin American countries took part in the registry between November 2017 and November 2019. A total of 974 cases were uploaded to the platform (tier 1), of which 761 were approved and included in the final analysis; 189 cases were rejected by country coordinators (tier 2) and 24 cases were rejected by the ALAT central committee (tier 3), and were therefore excluded from the final analysis.
Of the total number of patients included, 569 (74.7%) were men with a mean age of 71.9 (±8.3) years. Approximately half (52.3%) were former smokers. The median time from onset of symptoms to diagnosis was 12 months (IQR 6–24); no differences were found between participating countries (Fig. 2). Median follow-up was 14 months (IQR: 5–30). The principal study variables are described in Table 1. Autoimmune tests showed that 23 (3%) patients were positive for RF and 80 (11%) for ANA, the fine speckled IIF pattern being the most frequent (42.5%). Specific IIF patterns were only described in 15 cases (8 cytoplasmic, 6 nucleolar, and 1 centromere). The following systemic signs and symptoms were observed: xerophthalmia 16/753 (2.1%), xerostomia 32/753 (4.2%), joint symptoms 29/753 (3.8%), muscle weakness 27/753 (3.6%), and Raynaud's phenomenon 6/753 (0.8%).

Baseline characteristics of the REFIPI cohort (n=761).
| Men, n (%) | 569 (74.7) |
| Age, years, mean (SD) | 71.9 (8.3) |
| Onset of symptoms to diagnosis in months, median (IQR) | 12 (6–24) |
| Dyspnoea Grade 0 mMRC, n (%) | 60 (7.9) |
| Grade 1 mMRC, n (%) | 228 (30.3) |
| Grade 2 mMRC, n (%) | 300 (39.8) |
| Grade 3 mMRC, n (%) | 133 (17.6) |
| Grade 4 mMRC, n (%) | 32 (4.2) |
| Non-smokers, n (%) | 341 (45.3) |
| Former smokers, n (%) | 394 (52.3) |
| Current smokers, n (%) | 18 (2.4) |
| Family history of fibrosis, n (%) | 63 (8.4) |
| ANA+, n (%) | 80 (11.1) |
| Rheumatoid factor+, n (%) | 23 (3.1) |
| PASP, median mmHg (IQR) (n=492) | 29 (24–39.5) |
| PASP>40mmHg, n (%) | 123 (25%) |
| Finger clubbing, n (%) | 315 (41.8) |
| Velcro crackles, n (%) | 732 (97.2) |
| GER symptoms, n (%) | 283 (37.6) |
| % FVC, mean (SD) [n=744] | 70.9 (19.8) |
| % DLCO, mean (SD) [n=477] | 53.7 (45.4) |
| Meters in 6MW, mean (SD) [n=497] | 380.8 (135.8) |
| HRCT typical UIP, n (%) | 511 (67.8) |
| HRCT possible UIP, n (%) | 230 (30.5) |
| HRCT inconsistent with UIP, n (%) | 12 (1.6) |
| Surgical lung biopsy, n (%) | 124 (16.3) |
| Histology typical UIP, n (%) | 114 (91.9) |
| Histology probable UIP, n (%) | 6 (4.8) |
| Histology possible UIP, n (%) | 4 (3.2) |
| Histology no UIP, n (%) | 0 (0) |
6MW: 6-minute walk test, ANF: antinuclear factor, DLCO: diffusing capacity for carbon monoxide, FVC: forced vital capacity, GER: gastroesophageal reflux, HRCT: high-resolution computed tomography, IQR: interquartile range RF: rheumatoid factor, UIP: usual interstitial pneumonia, SD: standard deviation.
In the respiratory function test, the average baseline FVC and DLCO was 70.9% and 53.7%, respectively. No differences in baseline FVC % were observed among countries (Fig. 3).

Regarding treatment (Table 2), 553 patients (72%) received at least 1 antifibrotic drug, 375 (67.8%) received pirfenidone, 103 (18.6%) received nintedanib and 75 (13.5%) received both antifibrotics in succession. Regarding other treatments, 24 (4.15%) patients received triple anti-inflammatory/immunosuppressive therapy (azathioprine+corticosteroids+N-acetylcysteine) and 337/573 (58.8%) received anti-gastro-oesophageal reflux treatment.
Pharmacotherapy.
| Triple therapya, no. (%) | 24/578 (4.2) |
| Antifibrotic therapy, n (%) | 553/761 (72) |
| Pirfenidone, n (%) | 375/553 (49.3) |
| Pirfenidone dose mg, median (IQR) | 2000 (1800–2403) |
| Pirfenidone treatment in months, mean (SD) | 27 (3.6) |
| Nintedanib, n (%) | 103/553 (13.5) |
| Nintedanib dose mg, mean (SD) | 283.6 (42.9) |
| Nintedanib treatment in months, mean (SD) | 10.9 (8.2) |
| Pirfenidone+nintedanib (successive) | 75/553 (13.5) |
| GER therapy, n (%) | 327/573 (57) |
| Proton pump inhibitor, n (%) | 278 (82.5) |
| Anti H2, n (%) | 57 (16.9) |
| Prokinetics, n (%) | 2 (0.6) |
GER: gastroesophageal reflux, IQR: interquartile range, SD: standard deviation.
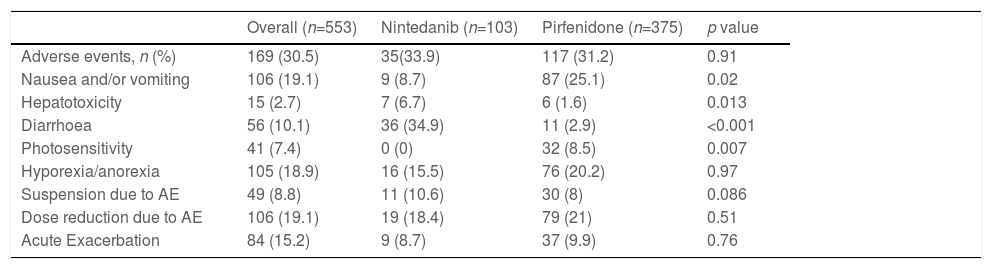
Antifibrotic-related adverse events were observed in 169 patients (30.5%), 117 (31.2%) of which were related to pirfenidone and 35 (33.9%) to nintedanib. The comparative analysis showed no statistically significant difference between nintedanib and pirfenidone in terms of the proportion of patients with adverse events (Table 3). Logistic regression analysis showed no association between adverse events adjusted for various variables, such as age, sex, baseline FVC %, baseline DLCO %, or smoking.
Adverse events associated with antifibrotics.
| Overall (n=553) | Nintedanib (n=103) | Pirfenidone (n=375) | p value | |
|---|---|---|---|---|
| Adverse events, n (%) | 169 (30.5) | 35(33.9) | 117 (31.2) | 0.91 |
| Nausea and/or vomiting | 106 (19.1) | 9 (8.7) | 87 (25.1) | 0.02 |
| Hepatotoxicity | 15 (2.7) | 7 (6.7) | 6 (1.6) | 0.013 |
| Diarrhoea | 56 (10.1) | 36 (34.9) | 11 (2.9) | <0.001 |
| Photosensitivity | 41 (7.4) | 0 (0) | 32 (8.5) | 0.007 |
| Hyporexia/anorexia | 105 (18.9) | 16 (15.5) | 76 (20.2) | 0.97 |
| Suspension due to AE | 49 (8.8) | 11 (10.6) | 30 (8) | 0.086 |
| Dose reduction due to AE | 106 (19.1) | 19 (18.4) | 79 (21) | 0.51 |
| Acute Exacerbation | 84 (15.2) | 9 (8.7) | 37 (9.9) | 0.76 |
AE: adverse events.
During follow-up, 84/746 patients (11.2%) presented an acute exacerbation, 38 (45.2%) of which died as a result. The usual treatment used for acute exacerbation was corticosteroids in 76 patients (90.5%), and corticosteroids and immunosuppressants in 2.4% of patients. No specific treatment was reported in 6 patients (7.1%). Thirty-three patients (4.3%) were included in the lung transplant list and only 11 (1.4%) were transplanted during the study period: 1 in Argentina, 1 in Mexico and 9 in Chile. A total of 88 (11.5%) deaths were reported over the 2 years of the registry, all of them related to IPF – 38 (43%) due to acute exacerbation and 50 (57%) to disease progression.
DiscussionREFIPI is the first Latin American IPF registry, and so far one of the largest multinational registries in terms of participating countries and number of patients. One hundred and forty six doctors from 14 different countries participated in this first stage of the project, and 761 patients were included in the final analysis (Fig. 4). The 3-tier verification system used to confirm diagnosis and central analysis of all the chest CT scans adds robustness to the study and increases the diagnostic certainty of IPF. This system allowed us to exclude 111 patients (11.4%) with an alternative diagnosis at the 2nd and 3rd tiers, and highlights the difficulties involved in diagnosing IPF in Latin America, even among specialists.

The REFIPI supplements the results reported in other studies and randomized clinical trials and, like other registries, includes a higher proportion of elderly male patients. On physical examination, 97.2% of patients were found to have Velcro crackles, highlighting the importance of this sign as a tool for the early detection of IPF.16 It is interesting to note that 8.4% of patients included in the REFIPI had a family history of pulmonary fibrosis. Although this finding was not corroborated by genetic studies, it suggests that about 1 in 10 patients with apparently sporadic IPF may have familial pulmonary fibrosis.
The main diseases included in the differential diagnosis of IPF are fibrotic hypersensitivity pneumonitis and autoimmune diseases. In terms of the latter, 10% of patients tested positive for autoantibodies, and only 15 presented a specific IIF ANA pattern; however, our 3-tier verification system confirmed that none of these cases met the criteria for a defined autoimmune disease or an interstitial disease with autoimmune features.17 Fibrotic hypersensitivity pneumonitis may often be indistinguishable from IPF,18 and is mainly diagnosed on the basis of inhaled antigens, the imaging pattern, and bronchoalveolar lavage tests.19,20 In the REFIPI, 12% of patients reported some exposure to inhaled antigens, mainly avian. All these cases, however, presented a typical or probable pattern of UIP on HRCT, and this, in the opinion of a multidisciplinary team, formed the basis for a reliable definitive or provisional diagnosis in each case. In terms of the HRCT pattern, only 12 (1.6%) patients presented a pattern inconsistent with UIP, but in all these cases lung biopsy was consistent with UIP.
Surgical lung biopsy was performed in 124 patients (16.3%) – less than the percentage found in most registries, such as the German (34.1%) and EurIPFreg (32%) registries.4,9,13 This could be due to the fact that these European registries were carried out prior to publication of the Fleischner Society20 recommendations, and the guidelines in force at that time recommended that patients with possible UIP on HRCT should undergo surgical biopsy for a firm diagnosis. In our study, surgical lung biopsy was performed on all patients who presented HRCT findings inconsistent with UIP, and in 112 of 230 patients (48.7%) who presented HRCT findings of possible UIP. This shows that half the time, specialists in Latin America do not perform lung biopsy on patients with possible UIP due to an exposure history within the appropriate clinical context, and rely instead on a provisional high confidence diagnosis (70%-90%) for IPF, as recommended by the Fleischner Society.21
Moreover, most of the pulmonologists participating in the REFIPI are affiliated with the ILD section of ALAT and specialise in treating patients with this type of disease (participation bias). This could have led them to include mostly patients with a firm clinical diagnosis.
Regarding the delay in diagnosis, patients had experienced symptoms for a median of 12 months before diagnosis. This period, however, varied widely among participating countries, and is evidence of the considerable delay that characterizes the diagnosis of this disease. Nevertheless, it is interesting to note that our median delay was shorter than that observed in most multinational registries published to date. For example, in the eurIPFreg12 and EMPIRE9 registries, mean time from onset of symptoms to diagnosis was 21.8 and 19.2 months, respectively.10,13 In the REFIPI, the delay in diagnosis was similar to that reported in the United States registry (IPF-PRO), namely 13.6 months,5 although in half of all registry patients the delay was greater than 1 year. Delays in the diagnosis of IPF have been associated with worse survival, and our results together with those reported in other regions underscore this problem of vital global importance.22
In terms of treatment, it is important to draw attention to the low proportion of patients who were treated with corticosteroids and immunosuppressants (4.2%). This treatment has been associated with a higher rate of hospitalization and mortality in patients with IPF.23 Similar results emerged from the EurIPFreg registry where less than 10% of patients received anti-inflammatory/immunosuppressive treatment over the same period as the REFIPI.13 In contrast our results, the German INSIGHT registry contained a high proportion of patients (26.1%) receiving corticosteroids.4,24 The same was true of a survey conducted in France and several randomized controlled clinical trials.25
A significant proportion of patients (72%) in the REFIPI were receiving some type of antifibrotic drug. This could be due to the aforementioned participation bias, as all participating pulmonologists were affiliated with the ILD section of ALAT and, given their extensive experience with IPF, would have been more likely to prescribe antifibrotics to their patients. Most of these patients received pirfenidone (49.3%) compared to nintedanib (13.5%). This is understandable in Latin America, where pirfenidone was marketed before nintedanib in most countries in the region. The same trend was observed in the Spanish registry, where 51.9% of patients received pirfenidone versus 17.9% receiving nintedanib, one reason being that pirfenidone was available 2 years prior to nintedanib in Spain.9
The same pirfenidone- and nintedanib-related adverse events as described in the initial ASCEND22 and INPULSIS/TOMORROW26–28 clinical trials were observed in the REFIPI, although in a lower proportion, insofar as adverse events were only reported in 31.2% and 33.9% of patients taking pirfenidone and nintedanib, respectively. The most common adverse events associated with pirfenidone were nausea, hyporexia/anorexia and photosensitivity in 25.1%, 20.2% and 8.5%, respectively. In the case of nintedanib, the most frequent adverse events were diarrhoea and hyporexia in 33.9% and 15.5% of patients, respectively.
The SEPAR registry also describes a lower rate of adverse events (23.4%), although the proportion of patients with nausea, anorexia and photosensitivity (9.5%, 7.8% and 5.7% respectively) was lower than that reported in REFIPI.9
Interestingly, the proportion of patients with photosensitivity in REFIPI (8.5%) and in the Spanish SEPAR registry8 (5.6%) was significantly lower than that found in the ASCEND (28.1%) study, even though a significant proportion of Latin American countries are located in latitudes with a high ultraviolet radiation index. This finding could be due to the different phototypes found in our region and a more widespread use of sunscreen.25
In the TOMORROW/INPULSIS trials investigating nintedanib, 61.5%, 11.2%, and 24.3% of patients reported diarrhoea, anorexia, and nausea, respectively.27,28 In the REFIPI, we observed a significantly lower rate of diarrhoea (33.9%). Although this could be underestimated due to the different study methodologies used, similar results were found in other real-world experience studies with nintedanib.29
Finally, only 8% of our population receiving pirfenidone and 10.6% receiving nintedanib had to suspend treatment due to adverse events. This contrasts with the findings of the pivotal ASCEND and INPULSIS/TOMORROW clinical trials, in which treatment had to be suspended in 14.4% and 20.6% of cases, respectively.26–28 The same trend has been reported in other international registries, and shows that both drugs are well-tolerated in clinical practice.
The REFIPI has both strengths and weaknesses. On the one hand, it is the first multinational, unsponsored, Latin American IPF registry, and has the backing of a recognized scientific society (ALAT). The 3-tier verification system and independent HRCT pattern analysis increases the level of confidence in the diagnosis of IPF. A large number of specialists from many different countries participated in the REFIPI, making it a good example of collaboration and a true representation of IPF in Latin America. The results of the REFIPI have given us the chance to identify and correct some major problems in the management of patients with IPF in Latin America: misdiagnosis of IPF (11.4% of cases were excluded for this reason), delay in diagnosis (12 months), and poor access to lung transplantation (only 1.4% of patients were transplanted).
This study has limitations that are typical of any registry, namely, case selection bias and intervention bias on the part of participating physicians. The number of cases contributed by each country also differs significantly, and is proportional to the number of inhabitants (Fig. 4). This is mainly due to lower adherence to the registry in some countries and/or delays in joining the REFIPI due to delays by some institutional review boards in approving the registry protocol. Finally, it is important to clarify that the REFIPI is not a prevalence study but a real-world observational study of data from IPF patients submitted voluntarily from countries across Latin America.
ConclusionsThe REFIPI is the first Latin American IPF registry. Like other registries, we observed difficulties and excessive delays in IPF diagnosis in Latin America. Most patients in the REFIPI received antifibrotics, which were well tolerated and associated with a lower rate of adverse events than reported in clinical trials.
FundingNone.
Conflict of interestsThe authors have no conflict of interest to declare.
Our thanks go to: Dr. Alberto Hegewish for providing the digital platform for uploading patient data; Daniel Pereira (ALAT web master) for his help in designing the REFIPI web platform and maintaining it throughout the study period; the members of the ALAT executive committees since 2014 for the support given to the Interstitial Lung Diseases section; and Dr. Pablo Donati for devoting his time to performing an excellent statistical analysis.
Our sincere and heartfelt thanks also go to all ALAT pulmonologists for their ongoing support, and for having transformed our “utopian” REFIPI project, into a reality for Latin America.
Argentina:
Alchapar Ramón Ángel (Medicina Respiratoria. Mendoza, Argentina), Alonso Álvaro Santiago (Punto Norte Rehabilitación Respiratoria. Buenos Aires, Argentina), Aruj Patricia (Instituto de Investigaciones Médicas “Alfredo Lanari”. Buenos Aires, Argentina), Azcona María Susana (Hospital Escuela de Agudos Ramón Madariaga. Posadas, Argentina), Baillieau Nicolás Augusto (Clínica 25 de Mayo. Mar del Plata, Argentina), Baldasaria Roque Antonio (Hospital Centro de Salud “Zenon J. Santillan”. Tucumán, Argentina), Balla Noelia (Hospital de la Baxada Dra. Teresa Ratto, Paraná, Entre Ríos, Argentina), Bragado Lucas Agustín (Hospital Formenti El Calafate. Santa Cruz, Argentina), Cervantes Cecilia (Hospital Italiano. Cordoba, Argentina),Cuestas Érica (Sanatorio Allende Cerro. Córdoba, Argentina), Elías Marcos (Hospital Privado de Córdoba. Cordoba, Argentina), Donati Pablo (Facultad de Ciencias Veterinarias de la Universidad de Buenos Aires, Catedra de anestesiología y algiología. Buenos Aires, Argentina), Fassola Leandro (Hospital de Rehabilitación Respiratoria “María Ferrer”. Buenos Aires, Argentina), Gamarra Antonella (Hospital Italiano de Córdoba. Córdoba, Argentina), García Jorge Andrés (Sanatorio Allende. Córdoba, Argentina), Gil Beatriz Liliana (Hospital Regional Concepción. Tucumán, Argentina), Goffredo Diego Hernán (Hospital Regional de Comodoro Rivadavia. Chubut, Argentina), Lardizabal Ayelen (Hospital San Juan de Dios de La Plata. Buenos aires, Argentina), Leiva Sebastian (Hospital de Clínicas Virgen de Fátima. La Rioja, Argentina), Lisanti Raúl (Instituto de Investigaciones Respiratorias. Mendoza, Argentina), Mannarino Silvina (Hospital de Emergencias Clemente Álvarez. Rosario, Argentina), Maritano Furcada Joaquín (Hospital Italiano de Buenos Aires. Buenos aires, Argentina), Martinez Gustavo (Consultorio Privado de Neumonología de San Fernando del Valle de Catamarca. Catamarca, Argentina), Masdeu Martin (Hospital Pirovano. Buenos Aires, Argentina), Molinari Luciana (Clínica Roca de General Roca. Rio Negro, Argentina), Nasca Diego (Consultorio privado de la ciudad de Lules, Tucumán, Argentina), Naval Norma (Hospital AC Padilla. Tucumán, Argentina, Nevado Alberto Jorge, Otaola María (Instituto de Rehabilitación Psicofísica (IREP). Buenos Aires, Argentina), Papucci Tulio (Instituto Neumonológico del sur (INeuS) de Bahía Blanca. Buenos Aires, Argentina), Paulin Francisco (Hospital Fernández. Buenos Aires, Argentina), Penizzotto Miguel (Sanatorio San Roque de Curuzú Cuatiá. Corrientes, Argentina), Piumatti Fernando (HIGA Dr. José Pena. Bahía Blanca. Argentina), Renom Horacio Alfredo, Rivera Luis Tomás, Robles Adriana (Hospital San Bernardo. Salta, Argentina), Rossi Pamela (Hospital Santojanni. Buenos Aires, Argentina), Sabas Mario Fernando (Hospital Oscar Allende. Mar del Plata, Argentina), Sebastiani Javier (Clínica Pueyrredon de Mar del Plata. Buenos Aires, Argentina), Sheridan Lucas (Centro de Educacion Médica e Investigaciones Clínicas (CEMIC), Buenos Aires, Argentina and Clinical Consultant to Boehringer Ingelheim since February 2019. Buenos Aires, Argentina), Sparvoli Fabricio Pablo (Hospital Gomendio de la Ciudad de los Arroyos. San Nicolás de los Arroyos, Buenos Aires), Susini María de los Milagros (Hospital Escuela de Corrientes. Corrientes, Argentina), Uribe Echevarria María Elisa (Hospital Italiano de Córdoba. Córdoba, Argentina), Usandivaras Marcela (Sanatorio 9 de Julio. Tucumán, Argentina), Valdez Maximiliano (Hospital Neumonológico Gumercindo Sayago. Santiago del Estero, Argentina), Welker Gustavo Alejandro (Hospital Privado de Rosario), Zabert Ignacio (Universidad Nacional del Comahue. Neuquen, Argentina).
Bolivia:
Cortez Olivera Noemi Justina (Hospital de la Caja de Salud de la Banca Privada. La Paz, Bolivia), López López Antonio Gonzalo (Hospital Elizabeth Seton. Cochabamba, Bolivia).
Brazil:
Fukuda Cesar Yoshito (Universidad Federal de Sao Pablo. Sao Pablo, Brazil) Kawano Leticia (Instituto de Pesquisa do HCor, Hospital do Curazao, San Pablo, Brazil), Lima Silva Mariana (Instituto de asistencia medica ao Servidor Publico Estadual-Lamspe. San Pablo, Brazil), Rodríguez Silvia Carla Sousa (Instituto de asistencia medica ao Servidor Publico Estadual-Lamspe. San Pablo, Brazil), Messias Lúcia Helena (Hospital Universitario Joâo de Barros. Belém, Brazil), Guilmar Alves Zonzin (UNIFOA. Volta Redonda, Brazil).
Chile:
De la Fuente Mandiola Isabel (Hospital Dipreca. Santiago, Chile), Delgado Diemen (Asociación Chilena de Seguridad. Santiago, Chile), Glasinovich Valezca (Clínica Indisa, RedSalud Vitacura. Santiago, Chile) Pozo Valeria (Consultorio Privado de Neumonología. Concepción, Chile), Reyes Felipe (Instituto Nacional del Tórax. Santiago, Chile), Salinas Mauricio (Instituto Nacional del Tórax. Santiago, Chile), Sepulveda Claudia (Instituto Nacional del Tórax. Santiago, Chile), Vargas Manuel (Hospital Ernesto Torres Galdames. Indique, Chile), Velásquez José Luis (Hospital San Juan de Dios. Santiago, Chile).
Colombia:
Cano Diana Jimena (instituto neumológico del oriente. Bucaramanga, Colombia), Celis Carlos Andrés (Hospital Universitario San Ignacio. Bogotá, Colombia), Duran Mauricio (Fundación Neumológica Colombiana. Bogotá, Colombia), Galindo Javier Leonardo (Hospital Universitario Mayor Méderi. Bogotá, Colombia), González García Mauricio (Fundación Neumológica Colombiana. Bogotá, Colombia), Rincon Alvarez Emily (Fundación Neumológica Colombiana. Bogotá, Colombia), Rodriguez Camilo Andres (Fundación Neumológica Colombiana. Bogotá, Colombia)
Ecuador:
Calle Delgado Catalina Alexandra (Hospital Axxis. Quito, Ecuador), Jaramillo Enrique Feliciano, Loor Shirley Mariuxi, Mendoza Bosco Fabián, Torrachi Carrasco Aldo Mateo (Universidad del Azuay. Cuenca, Ecuador)
Guatemala:
González Velásquez Edilzar (Centro de Estudios Pulmonares. Guatemala, Guatemala)
Mexico:
Alemán Marquez Ángel (Hospital Naval de Veracruz. Veracruz Mexico), Caroline Armeaga (AGProsalud, Ensenada Baja California, Mexico), Arreola Morales Alejandro (General Hospital Dr. Columba Rivera Osorio. Ciudad de Pachuca de Soto, Mexico), Barajas Ugalde Daniel (Hospital de Especialidades Centro Medico Nacional de Occidente IMSS. Guadalajara Mexico), Bazaldua Pearl (CREPID, Mexico City, Mexico), Benitez Geovanni (Instituto Nacional de Enfermedades Respiratorias “Dr. Ismael Cosio Villegas. Mexico City, Mexico), Chavarría Carlos (Centro Médico de Tijuana, Baja California, Mexico), Espinoza Manuel (Centro Médico de Especialidades, Ciudad Juarez, Chihuahua, Mexico), Estrada Garrido Andrea (Instituto Nacional de Enfermedades Respiratorias “Dr. Ismael Cosio Villegas. Mexico City, Mexico), Grave Sergio Manuel (Hospital General de Ensenada, Baja California, Mexico), Guillen Fernando (Hospital Dr. Belisario Domingez. Chiapas, Tuxtla Gutierrez, Mexico), Herrera García José Carlos (Hospital Ángeles Puebla. Puebla, Mexico), Mateos Heidegger (Instituto Nacional de Enfermedades Respiratorias “Dr. Ismael Cosio Villegas. Mexico City, Mexico), Mayorga Reyes Jorge Alain (Hospital Christus Muguerza Reynosa. Reynosa, Mexico), Medina Ivan (Star Medica Luna Park, Mexico State, Mexico), Morales Sanchez Lisvenia (Hospital General de Chilpancingo Dr. Raymundo Abarca Alarcón. Guerrero, Mexico), Padua José (Centro Médico ABC, Mexico City, Mexico), Rojas Soledad Ariel (Hospital Militar el Chivatito. Mexico City. Mexico), Sánchez Nestor Ulises (Hospital Regional ISSSTE, León Guanajuato, Mexico), Sánchez Gutiérrez Jaime Alejandro (Instituto Nacional de Enfermedades Respiratorias “Dr. Ismael Cosio Villegas. Mexico City, Mexico), Santiago Luis Alberto (Hospital de Especialidades Médicas Santa Fé, Tapachula, Chiapas, Mexico), Sarmiento Gonzalo (Humanitas Hospital de Tlaxcala, Tlaxcala, Mexico), Toledo Javier (Hospital Puebla, Puebla, Mexico), Torres Alan (Clínica del Centro, Chihuahua, Mexico), Zaldivar Crosby Guillermo (Hospital General de Zona Numero 6. Nuevo León, Mexico), Zuloaga Roman Yris Dianora (Hospital General ISSSTE Saltillo. Saltillo, Mexico.
Panama:
Guevara Eric (Consultorios América, Ciudad de Panama, Panama), Hevia Quiróz Eduardo Gabriel (Clínica Hevia. Panama City, Panama), Jaramillo Fabio (CEDITER, Panama City, Panama). Panama City, Panama), Jiménez Raúl (CEDITER, Ciudad de Panama, Panama), Marquez Fernando (CEDITER, Panama City, Panama), Perea Tarsicio (Instituto de Neumología y Alergias INASA. Panama City, Panama).
Paraguay:
Benitez Ayala Silvio Gabriel (Instituto Nacional de enfermedades respiratorias y del ambiente. Asunción, Paraguay), González Fernando Ramón (Instituto Nacional de Enfermedades Respiratorias y del Ambiente “Juan Max Boetner” (INERAM). Asunción, Paraguay), Medina Diego (Instituto de Previsión Social (IPS). Asunción, Paraguay), Perez Bejarano Domingo (Hospital General de Luque. Luque, Paraguay).
Peru:
Azañero Lujan Mario, Chavez Bazán Tania, Giron Atoche Vicente Angel (Hospital Nacional Arzobispo Loayza. Lima, Peru) Herrera Flores Edwin (Hospital Nacional Arzobispo Loayza. Lima, Peru), Jaramillo Peralta Isabel Bertha (Hospital Nacional Arzobispo Loayza. Lima, Peru), Venero Caceres María del Carmen (Hospital Nacional Arzobispo Loayza. Lima, Peru), Tania Homaira Chávez Bazan. (Hospital Belén de Trujillo. Trujillo, Peru), Shanery Norma Gonzales Vargas (Hospital Nacional Arzobispo Loayza. Lima, Peru).
Dominican Republic:
Matos Comas Candy Yamile (MedicalnetB. Santo Domingo, Dominican Republic), Nuñez Sabrina Isabel (Clinica Independencia de Santo Domingo, República Dominicana), Jaquez Miguelina (Hospital Metropolitano de Santiago. Santiago, Dominican Republic), Mallol Anandy (Instituto Materno Infantil. Santiago, Dominican Republic), Garcia Batista Natalia (Clínica Universitaria Unión Médica del Norte. Santiago, Dominican Republic), Castillo Joel (Clínica Corominas. Santiago, Dominican Republic)
Uruguay:
Lapiedra Jimena (Hospital Maciel. Montevideo, Uruguay), Rodríguez Cáceres Martha Inés (Hospital Maciel. Montevideo, Uruguay).
Venezuela:
Torrealba Carlos (Urológico San Román. Caracas, Venezuela).
The following are the supplementary data to this article:







