

















Definition
Diffuse interstitial lung disease (DILD) is a category comprising a series of entities with similar clinical, radiologic, and lung function presentations, in which the principal pathological alterations affect the interstitial alveolar structures. The term DILD does not, in fact, properly describe the underlying pathogenetic mechanism of these clinical entities because they very often affect not only the interstitial alveolar structures, but also the small airways and the pulmonary vasculature.1,2
Etiology and Classification
The etiology of DILD is extremely varied. More than 150 different causes are currently known, although identification of the causal agent is only possible in approximately 35% of cases. The classification of these entities has been modified recently following the publication of a consensus statement produced jointly by the American Thoracic Society (ATS) and the European Respiratory Society (ERS) (Table 1).3,4 Three groups of DILDs can be distinguished. The first group (idiopathic interstitial pneumonias) comprises a set of clinico-pathological entities whose histological definition has attracted a great deal of attention in recent years. The second group includes the DILD of known causes and those associated with other well-defined disease entities. This group includes the pulmonary manifestations of the collagen diseases, the histology of which is often indistinguishable from that of the idiopathic interstitial pneumonias. This group also includes drug-induced DILD, and DILD caused by organic and inorganic agents (extrinsic allergic alveolitis [EAA] and pneumoconiosis respectively), and those associated with hereditary diseases. The third group is made up of a subset of disorders which, while they are idiopathic, present well defined clinical and histologic features.

Epidemiology
Epidemiological data on DILD are sparse. The incidence and prevalence reported in the literature are highly variable because the epidemiological methods used differ from one study to another. Moreover, following recent changes in the classification of DILD, it is impossible to obtain precise information on the incidence and prevalence of newly defined clinical entities, such as nonspecific interstitial pneumonia (NSIP). Furthermore, most DILD registers have been compiled on the basis of surveys that exclusively targeted pulmonology departments, so that they do not include the diseases diagnosed by other departments or types of hospital. It is, however, generally agreed that the most common DILD are idiopathic pulmonary fibrosis (IPF) and sarcoidosis, followed by EAA and those related to collagen disease.5
Diagnostic Methods
Medical History
Diagnosis can be achieved in one third of DILD cases solely on the basis of an accurate medical history.6,7
- Age and sex. In patients aged between 20 and 40, the most common disorders are sarcoidosis, histiocytosis X, lymphangioleiomyomatosis (LAM), and the DILD associated with collagen diseases. IPF is usually diagnosed in patients over 50 years old. LAM is found virtually only in women. The DILD associated with collagen disease are more common among women.
- Family history. This can provide very useful information. Patients with IPF may have a family member who is also affected by this disease (familial pulmonary fibrosis). Alveolar microlithiasis, tuberous sclerosis, neurofibromatosis, and sarcoidosis are other examples of clinical entities with hereditary history which are associated with DILD.
- Tobacco use. Certain DILD primarily affect smokers. These include desquamative intersitial pneumonia (DIP), respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), and histiocytosis X. The opposite is true of sarcoidosis and EAA, which occur more frequently in nonsmokers than smokers.
- Occupational and work history. EAA is caused by exposure to organic agents, and pneumoconiosis by exposure to inorganic dust. A complete occupational history should be recorded with details of work activities and any possible exposure to inorganic dust. The date and duration of any exposure should be noted. All work activities should be recorded in chronological order.
- Drug history. Drugs are a not uncommon cause of DILD. Consequently, all current and past medications taken by the patient should be noted, including details of the dose and duration of treatment.
- Radiotherapy. A history of chest radiation therapy could be the cause of DILD.
- Systemic diseases. Identify and investigate possible symptoms of the systemic diseases (collagen disease, sarcoidosis) that can be associated with DILD.
Clinical Signs and Symptoms
The most common symptoms are dyspnea on exertion and cough. The most prominent symptom is progressive dyspnea on exertion, which is often associated with abnormalities in the chest radiograph. Patients with normal chest radiographs may, however, complain of dyspnea, while in other cases the disease may be discovered in asymptomatic patients by way of a chest radiograph taken for some other reason. Onset of dyspnea is usually gradual but progressive, and for some time it may be the only symptom. As a result, patients tend to consult their physician weeks or months after onset of the disease, which delays diagnosis and treatment. Most patients present with dry cough. Hemoptysis is rare, but occurs occasionally in patients with LAM. Patients with coal worker's pneumoconiosis may present melanoptysis. The following diseases can cause acute or subacute respiratory symptoms, occasionally associated with fever and other systemic symptoms: NSIP, acute interstitial pneumonia (AIP), EAA, drug-induced pneumonitis, cryptogenic organizing pneumonia (COP), and pulmonary eosinophilias. Substernal or pleuritic chest pain is rare. Acute pleuritic pain caused by pneumothorax can be a sign of histiocytosis X or LAM.1,2,7
The most useful information obtained from the physical examination is the presence of crackles and finger clubbing, although these signs are not present in all DILDs. Wheezing would tend to suggest EAA or pulmonary eosinophilia. Apart from these general characteristics, each form of DILD has its own clinical peculiarities (see the specific characteristics of the different forms of DILD).
As DILD progresses, pulmonary hypertension may develop and lead to chronic cor pulmonale (edema, hepatomegaly, and jugular venous distension).
It is also essential to look for the presence of extrapulmonary signs and symptoms that might indicate any of the diverse diseases associated with DILD (Table 2).

Blood Tests
Blood test results are of interest in the diagnosis of certain DILD (Table 3).1,2,7

Sarcoidosis patients sometimes have raised serum angiotensin converting enzyme values. This parameter has been used as an activity marker in such patients, but with debatable results.8 In light of this, and taking into account that angiotensin converting enzyme levels may be raised in other DILD, this test should be used with caution in the diagnosis of DILD.
Radiographic Imaging
Chest radiography is still an indispensable tool in the initial radiographic assessment and subsequent monitoring of patients with DILD for various reasons: a) 90% of patients with DILD have radiographic abnormalities at the time of diagnosis; b) the distribution of the interstitial pattern and associated features is a useful diagnostic datum, and c) comparison of serial radiographs is useful for monitoring the course of the disease. The radiographic features associated with DILD are ground glass opacities; reticular, small nodular, and reticulonodular patterns; and honeycombing. These generally affect both hemithoraces diffusely and are accompanied by a reduction in the size of the lung fields. An alveolar pattern is indicative of the following DILD: AIP, NSIP, DIP, COP, lymphoid interstitial pneumonia (LIP), alveolar proteinosis, EAA, and pulmonary eosinophilias. The distribution of the pulmonary opacities and the presence of other radiographic abnormalities can point to a specific diagnosis6 (Table 4).

Computed axial tomography of the thorax is a more sensitive tool than chest radiography in the investigation of the abnormalities of the pulmonary interstitium. In any investigation of DILD a high resolution computed tomography (HRCT) scan should always be obtained. This imaging technique facilitates detection of the disease in patients with a normal chest radiograph. It is also useful in assessing the extent and nature of the parenchymal lesions because reticular patterns are indicative of fibrosis and ground glass patterns of inflammation. However, when ground glass patterns are found in conjunction with reticular patterns, they may be indicative of conglomerate masses rather than inflammation. In IPF, histiocytosis X, asbestosis, and LAM, HRCT findings are considered to be diagnostic criteria. In other clinical entities (COP, EAA, sarcoidosis, alveolar proteinosis), HRCT findings are helpful in reaching a diagnosis. HRCT scanning should also be used to identify the correct site for transbronchial and surgical biopsy, and bronchioalveolar lavage (BAL). A normal HRCT does not exclude a diagnosis of DILD.1,2,7
The clinical value of magnetic resonance imaging, despite the possibilities it offers in the investigation of the chest, is still purely speculative. Bone radiography can be useful in the diagnosis of collagen disease, sarcoidosis, and histiocytosis X.
Pulmonary scintigraphy using gallium-67 (67Ga) is not useful in the diagnosis of DILD owing to its lack of specificity and sensitivity. Whole body gallium imaging using 67Ga is useful in a few rare cases of difficult-to-diagnose sarcoidosis, since it reveals the 2 signs typical of the disease: a) the lambda (bilateral hilar and right paratracheal lymphadenopathy uptake), and b) the panda (uptake in the lacrimal and parotid glands). Although some authors have made a case for the usefulness of scintigraphy using 99Tc-DTPA (diethylene triamine pentaacetate) to assess epithelial permeability and the evolution of DILD, available data is too meager to recommend its use.
Lung Function Testing
Lung function testing is a key tool in reaching a diagnosis, determining prognosis, and in monitoring the course of the disease and the patient's response to treatment. In 15% of cases, lung function impairment is the first symptom of DILD. Nevertheless, a normal lung function test result does not exclude the possibility of DILD. A correlation exists between the degree of functional impairment and the degree of destruction of the lung parenchyma, although these tests do not provide any evidence as to whether the impairment is due to alveolitis or fibrosis. In forced spirometry, the functional pattern is characterized by a restrictive defect. The following DILD can be associated with an obstructive ventilatory pattern: EAA, sarcoidosis, histiocytosis X, eosinophilic pneumonia, and LAM. Total lung capacity and the different subdivisions of the lung volumes are reduced. In DILD associated with pulmonary emphysema, forced vital capacity (FVC), and pulmonary volumes are normal. The diffusing capacity of lung for carbon monoxide (DLCO) is reduced (this is one of the most sensitive indicators of DILD). The KCO (DLCO/alveolar volume transfer coefficient) is usually normal or moderately low. Arterial blood gasometry shows an increase in the P(A-a)O2 (alveolar-arterial gradient of oxygen) with moderate hypocapnia. Arterial hypoxemia only becomes evident in the advanced stages of the disease, and hypercapnia in the final stages. Exercise testing reveals a low exercise tolerance because of the dyspnea associated with hypoxemia caused by the exercise. The role of exercise testing in the diagnosis of DILD is limited to the detection of the disease in breathless patients with a normal chest radiograph and lung function.7 Although only limited experience is available, the 6-minute walk test has proved useful in evaluating the course of the disease.1,2,9,10
Bronchoalveolar Lavage
Cellular and immunocytochemical analysis of BAL is very useful in the diagnostic assessment of DILD. Biochemical analysis and measurement of immunoglobulins do not contribute any useful data. Mineralogical analysis is, however, useful in the diagnosis of pneumoconiosis. The performance of serial BAL has not been shown to be of any use in determining prognosis or monitoring response to treatment. BAL may preclude the need for lung biopsy in some DILD. In most cases, however, the role of BAL in the process will be to guide the diagnosis, supporting a provisional diagnosis or suggesting an alternative.11 The role of BAL in DILD is shown in Table 5.

Lung Biopsy
Histological analysis of the lung parenchyma is, in many cases, needed to reach a conclusive and specific diagnosis of DILD.1,2,6,7
Transbronchial biopsies performed using a fiberoptic bronchoscope can confirm diagnosis in the following types of DILD: sarcoidosis, EAA. histiocytosis X, amyloidosis, LAM, alveolar proteinosis, COP, pulmonary eosinophilia, and some types of pneumoconiosis. They are not, however, useful in the diagnosis of idiopathic interstitial pneumonias (except COP). The fact that the lung parenchyma is found to be normal does not rule out the possibility of DILD.
Surgical lung biopsy using minithoracotomy or video-assisted thoracoscope is indicated in all cases in which a specific diagnosis of DILD has not been obtained using the investigation techniques described above. The advantages of video-assisted thoracoscopy are the shorter operating time, fewer postoperative complications, and shorter length of stay in hospital. The decision to perform a biopsy should be evaluated on a case-by-case basis, since its appropriateness will depend on the patient's clinical status and on the value of the possible advantages in diagnosis and treatment. Biopsy sites should be predetermined on the basis of the HRCT results. Samples should be taken from at least 2 different sites, one with a pathological macroscopic appearance, and the other with a normal macroscopic appearance.
Transparietal needle biopsy should not be used because of the small size of the lung sample obtained with this technique and the high incidence of secondary pneumothorax.
Diagnostic Process
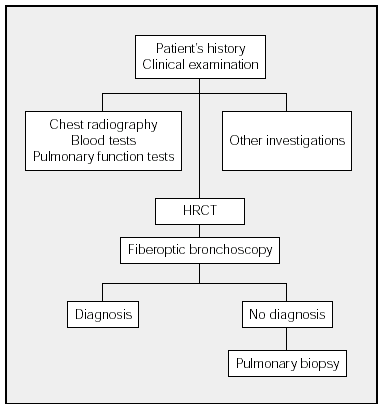
The diagnostic process of DILD is shown in the Figure. This process begins with history taking followed by a physical examination and chest radiography. The blood tests that should be ordered will depend on the differential diagnosis. The next step is to investigate respiratory function (spirometry, lung volumes, carbon monoxide transfer, and arterial gasometry). Exercise testing and the 6-minute walk test do not have to be performed in all cases of DILD. As a general guideline, these tests are indicated in IPF and the chronic forms of DILD that do not respond to initial treatment. The decision to undertake other investigations (such as eye examination and electrocardiogram, among others) will depend on the clinical picture and on the results of chest radiography and blood tests. Whenever possible, HRCT scans should be carried out before fiberoptic bronchoscopy. When a diagnosis has not been established, surgical lung biopsy should be used if possible.
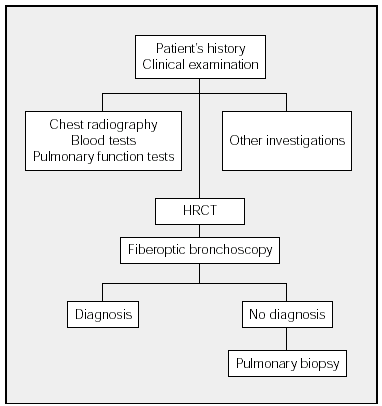
Figure. Diagnostic Process in Diffuse Interstitial Lung Disease. HRCT indicates high resolution computerized tomography.
Differential Diagnosis
A differential diagnosis should be drawn up when the clinical signs and radiographic picture are common to several clinical entities.1,2
- Heart failure. Pulmonary edema can give rise to a bilateral interstitial pattern. Heart failure should be suspected in patients with a history of heart disease, and in the presence of cardiomegaly, B Kerley lines, pleural effusion, or predominantly perihilar infiltrates in the chest radiograph.
- Bronchiectasis. Although an interstitial pattern in the chest radiograph is one sign of bronchiectasis, the clinical manifestations and HRCT results will differentiate between this diagnosis and DILD.
- Pneumonia. The clinical picture and radiographic findings in some cases of pneumonia can be confused with those of acute cases of EAA.
- Lymphangitis carcinomatosa. Chest radiograph reveals a bilateral interstitial pattern with B Kerley lines. The definitive diagnosis can be confirmed by the BAL and transbronchial biopsy findings.
- Pulmonary infiltrates in immunodeficient patients. Pulmonary infiltrates in patients with lowered immunity, primarily those caused by opportunistic pathogens, produce interstitial patterns in chest radiographs. A history of lowered immunity and a microbiological analysis of BAL will generally confirm diagnosis.
- Diffuse pulmonary hemorrhaging. Such hemorrhaging produces diffuse alveolar or alveolointerstitial patterns in the chest radiograph. Clinical signs (anemia, hemoptysis), BAL results (the presence of hemorrhagic fluid and/or hemosiderophages), and the presence of immunological abnormalities (neutrophil anticytoplasmic antibodies, antinuclear antibodies, basal antimembrane antibodies) are generally sufficient to differentiate these cases from DILD.
- Lipoid pneumonia. On rare occasions, lipoid pneumonia causes bilateral interstitial infiltrates. Diagnosis can generally be confirmed by BAL findings (staining of lipid vacuoles in the alveolar macrophages) and the findings of transbronchial or surgical lung biopsy.
- Miliary tuberculosis and miliary disease caused by Bacillus Calmette-Guérin. A miliary pattern on a radiograph is seen in 6% of tuberculosis patients. Hematogenous dissemination of the Bacillus Calmette-Guérin is observed in patients with urinary bladder cancer who receive topical treatments containing Bacillus Calmette-Guérin and present a miliary pattern in chest radiographs.
Complications
Complications develop more often in patients receiving prolonged treatment with corticosteroids and/or immunosuppressive therapy, and in patients with IPF or other forms of DILD that progress to pulmonary fibrosis.12
- Respiratory insufficiency. Respiratory failure is the cause of death in 40% of cases. In the advanced stages of the disease, a large number of patients present chronic respiratory insufficiency. Patients with IPF or other forms of DILD that have progressed to pulmonary fibrosis may present a clinical picture characterized by dyspnea, fever, and rapidly progressive acute respiratory insufficiency. Chest radiographs reveal alveolar patterns, and extensive ground glass opacities are apparent on HRCT scans. In 30% to 40% of cases, exacerbation of the disease is caused by respiratory infections. However, in most cases it is impossible to identify any exacerbating cause, and acute respiratory insufficiency is attributed to the rapid progression of the disease. Examination of the lung biopsy reveals diffuse alveolar damage or organizing pneumonia in addition to the abnormalities typical of the underlying disease. Some 90% of patients die, and neither mechanical ventilation nor treatment with high doses of corticosteroids has been shown to have any beneficial effect.13
- Respiratory infections. Traction bronchiectasis, impaired ciliary clearance, and treatment with corticosteroids or immunosuppressive agents all make patients susceptible to respiratory infection caused by usual or opportunistic pathogens. The increase in the incidence of pulmonary tuberculosis in patients with IPF and silicosis is noteworthy in this respect.
- Pulmonary hypertension. In the advanced stages of IPF and other DILD that progress to fibrosis, pulmonary hypertension and cor pulmonale develop in 70% of patients and are the cause of death in 30% of cases.
- Lung cancer. There is a high incidence of lung cancer in both IPF and asbestosis. The characteristics and frequency of the histological cancer types associated with these DILDs are similar to those found in the population in general.
- Pulmonary thromboembolism. Pulmonary thromboembolisms is responsible for the death of 3% to 7% of patients with DILD. Predisposing factors are inactivity owing to dyspnea, right cardiac failure, and the presence of associated lung cancer.
- Pneumothorax. Pneumothorax is uncommon and found in only 3.6% of cases. It is accompanied by rapid clinical deterioration and respiratory insufficiency. Generally it is not resolved by chest drainage because of the rigidity of the lung parenchyma, which prevents the lung from re-expanding.
- Mycetoma. Mycetoma is a complication that may occur in patients with sarcoidosis who have destructive fibrotic pulmonary lesions.
Treatment
The fundamental aims of treatment are to avoid exposure to the causal agent, to suppress the inflammatory component of the disease (alveolitis), and to treat the complications. The first objective is only feasible when the etiology of the disease is known. The elimination of alveolitis is the only therapeutic treatment for idiopathic DILD, since no antifibrotic drugs of proven efficacy are currently available. The drugs used are corticosteroids and immunosuppressants. Indications and duration of treatment vary depending on the form of DILD (see the section "Specific Characteristics of Different Forms of Diffuse Interstitial Lung Disease"). Patients with secondary pulmonary hypertension can benefit from oxygen therapy and vasodilators. Iloprost (an analog of prostaglandin I2) may be an effective treatment. A recent study has demonstrated that sildenafil produces pulmonary vasodilatation and improved gas exchange. However, there is no recommended strategy.1,2,7,14
Lung Transplant
Lung transplant is the last therapeutic option for the DILDs that progress to fibrosis and cause respiratory failure. Transplant candidates must fulfill the general criteria for eligibility that apply to any sort of lung transplant and must not present contraindications (Tables 6 and 7).15,16


There are over 120 types of DILD that progress to fibrosis, and it is, therefore, very difficult to identify the transplant window (the ideal moment for transplanting for each patient, neither too early, nor so late that the timing compromises the viability of the operation). There are no protocols that summarize the indications for lung transplant in the case of this very heterogeneous group of fibrosing diseases.17 For patients with IPF, the accepted indications for evaluating the possibility of a lung transplant include progressive dyspnea despite appropriate immunosuppressive therapy, and persistent hypoxemia during rest or on exertion, which is generally associated with a decrease in FVC on the order of 60% to 70% of the predicted value and a DLCO of less than 50% to 60% of predicted.18 However, these criteria may not be applicable to other types of pulmonary fibrosis because no reliable theoretical prognostic models exist. Moreover, we do not have any data on large groups of patients that could be used to demonstrate that such a transplant would increase survival, as has been demonstrated in the case of IPF.19
One aspect of DILD that should be considered is the possibility that the disease may recur after transplant. Recurrence has been reported in cases of DIP, alveolar proteinosis, LAM, and sarcoidosis. There is little experience with recurrence of these diseases and its clinical repercussions. In the case of sarcoidosis, recurrence has few clinical repercussions, and there is no evidence that it increases the risk of transplant rejection.20
Single lung is the preferred type of transplant for patients with DILD. Bilateral lung transplants should only be considered in cases where there is some doubt about the behavior of the remaining lung. Cardiopulmonary transplant is only indicated for young patients with right refractory cardiac failure, a very rare occurrence.
Monitoring the Course of the Disease and the Patient's Response to Treatment
With respect to the question of monitoring the course of the disease and the patient's response to treatment, the ERS and ATS have issued consensus statements defining criteria for sarcoidosis and IPF, which may be used in a general way for other DILD. As a general guideline, the following are recommended (Table 8):

Quarterly Checkup:
- Assessment of symptoms (in particular dyspnea, using validated scales). Questionnaires on quality of life have received little attention in connection with DILD. Preliminary studies indicate that the most useful of these are the WHOQOL-100 (World Health Organization Quality of Life) and the SF-36 (36-Item Short-Form Questionnaire).21
- Chest radiograph.
- Lung function test (forced spirometry, lung volumes, DLCO, and resting arterial gasometry).
Annual checkup:
- HRCT.
- Exercise testing. Although they provide very valuable information, exercise tests are, in general, not routinely used to monitor response to treatment. In the experience of several authors, these tests can only be performed in 30% to 40% of cases.22 The ATS/ERS consensus is that an increase in PaO2 of more than 4 mm Hg during exercise is indicative of an improvement, and that a rise in P(A-a)O2 of more than 4 mm Hg is indicative of deterioration.3 Experience with the 6-minute walk test is scant, although its use is recommended.9,10
Deterioration in lung function, clinical status, or radiographic findings in patients with DILD indicates progression of the disease or lack of response to treatment, although such results could also be an indication of complications (see the section "Complications").
Specific Characteristics of the Different Forms of DILD
Idiopathic Pulmonary Fibrosis
IPF is a form of DILD characterized by the presence of a usual interstitial pneumonia (UIP) pattern in the histological examination of the lung parenchyma. It is estimated that the prevalence of the disease is 20 cases per 100 000 for males and 13 cases per 100 000 for females, making it the most common form of DILD. Its etiology is unknown, although it is probable that IPF is the consequence of the action of external agents in individuals with a genetic predisposition.23,24
Clinical features. The patient's age at the time of presentation of the disease is usually greater than 50 years. Onset is insidious, taking the form of progressive dyspnea and dry cough. The presence of constitutional symptoms should lead physicians to suspect an alternative diagnosis. On physical examination, Velcro-type crackles are found in 90% of cases and digital clubbing in 20% to 50% of cases. Blood tests may reveal abnormalities in markers of inflammation (C-reactive protein, globular sedimentation velocity, gamma globulins). Positive anti-nuclear antibodies or rheumatoid factor occur in 10% to 20% of cases, although the titers are low. Familial pulmonary fibrosis is a form of pulmonary fibrosis that affects 2 or more members of the same family. The characteristics of the disease are similar to those of the nonfamilial form, although the disease is usually diagnosed at a younger age.25
Chest radiography reveals reticular opacities, sometimes associated with honeycombing, with a basal and bilateral distribution. Alveolar patterns are rare, and their presence should alert the physician to the possibility of an alternative diagnosis. HRCT reveals a series of characteristic abnormalities that are considered to be diagnostic criteria. Their diagnostic sensitivity has been estimated at 90%. These abnormalities are as follows: reticular patterns, irregular septal thickening, traction bronchiectasis, and bibasilar, subpleural, and symmetrical honeycombing. HRCT findings are not considered to be diagnostic in the presence of parenchymatous micronodules, small bronchovascular nodules, or extensive ground glass opacity. HRCT abnormalities correlate with functional abnormalities.22 The abnormalities usually found in BAL are neutrophilia, sometimes in association with moderate eosinophilia. Lymphocytosis is not a characteristic feature of IPF, and in patients with lymphocyte counts of over 15% or eosinophil counts of over 20% other possible alternative diagnoses should be excluded.4
Diagnosis. A definitive diagnosis of IPF requires the presence of the histologic pattern of UIP. A set of criteria have been established that allow diagnosis with a sensitivity of over 90% when lung biopsy samples are not available (Table 9).3,4 The diagnosis of IPF is clinicopathological. It should be emphasized that a simple finding of a UIP pattern on histological examination of the lung parenchyma is not synonymous with a diagnosis of IPF. This histological pattern can also be associated with other clinical entities (Table 10).


Prognosis. This disease has a bad prognosis. Around 50% of patients die within 3 to 5 years of diagnosis.26
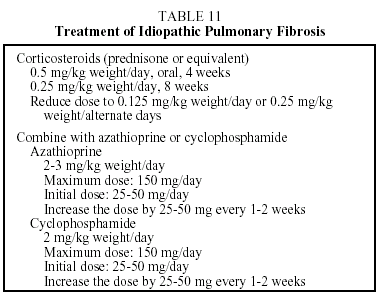
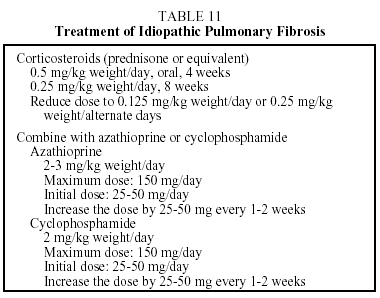
Treatment. No therapy has been found which modifies the prognosis of this disease. The only therapeutic strategy shown to be useful in slightly increasing survival is combined treatment with corticosteroids and azathioprine. The ATS/ERS have established guidelines concerning the therapeutic regimen that should be used (Table 11).3 This takes the form of the corticosteroid therapy administered in combination with cyclophosphamide or azathioprine. Azathioprine is more frequently used than cyclo-phosphamide because it provokes fewer side effects. When corticosteroids are contraindicated, the patient may be treated with azathioprine or cyclophosphamide. Colchicine, an antifibrotic agent, may provide an alternative for patients with a poor tolerance of corticosteroids or immunosuppressive therapy. The recommended dose is 0.6 to 1.2 mg per day. The duration of treatment depends on the course of the disease, but maintaining the initial regimen for at least 6 months is recommended. If the patient's condition is found to have improved or stabilized, the same regimen should be continued. If the patient's condition has deteriorated after 6 to 12 months of treatment, the possibility of a lung transplant should be considered. Some patients remain stable for long periods without treatment. These are patients with few or slight symptoms and only slight lung functional abnormalities. In such cases, until effective antifibrotic pharmaceuticals become available, the physician may decide not to start treatment until clinical or functional changes that indicate progression of the disease are noted.27 A recent study demonstrated a significant improvement in lung function in a small group of patients treated with interferon γ-1b.28 Studies are currently under way to confirm the efficacy of this drug, and preliminary results have shown an increase in length of survival.0
Nonspecific Interstitial Pneumonia
This is a clinicopathological entity first described in 1994 which comprises a group of interstitial lung disorders that present with pathological abnormalities not characteristic of other forms of idiopathic interstitial pneumonia. Until a few years ago, NSIP was included under the generic term IPF, although it is now known that it is an entirely differentiated entity.4,29
Clinical features. Onset of the disease is insidious or subacute, with cough and exertional dyspnea. Some 50% of patients present constitutional symptoms and some 30% finger clubbing. NSIP can be idiopathic (60% of cases) or drug-induced, or it can be associated with collagen disease, EAA, or a history of acute respiratory distress syndrome. Chest radiography and HRCT reveal nonspecific, nondiagnostic characteristics. These are symmetrical and basal ground glass opacities, sometimes associated with reticular patterns. Honeycombing is rare. Occasionally, HRCT findings in NSIP may be identical to those associated with IPF. BAL findings are variable and are not diagnostic. In some cases, both UIP and NSIP patterns have been observed in the same patients, which has led some authors to consider NSIP to be a precursor to UIP.
Two sub-groups of NSIP disorders have been defined according to pathological findings: Group I, primarily with inflammation; and Group II, primarily with fibrosis.23
Diagnosis. This should be established by means of surgical lung biopsy.
Prognosis. This depends on the degree of inflammation observed in the biopsy samples. In any case, the prognosis for patients with NSIP is more favorable than the prognosis for those with IPF.29
Treatment. Oral corticosteroids (prednisone or equivalent) at a dosage of 1 mg/kg of weight (maximum 80 mg) for one month. The dose is then reduced by 10 mg every 15 days until reaching 20 mg. This dose is maintained for 2 weeks, after which it is reduced to between 5 mg and 10 mg administered on alternate days, a regimen that is maintained until clinical resolution and stabilization of respiratory function parameters is achieved. If the patient does not respond to corticosteroid treatment, azathioprine is added using the dosage regimen recommended for IPF.3
Cryptogenic Organizing Pneumonia
The ATS/ERS consensus statement recommends the use of the term cryptogenic organizing pneumonia (COP) rather than the classic term bronchiolitis obliterans organizing pneumonia (BOOP) for two reasons: firstly, organizing pneumonia is the principal pathological characteristic of the disease; and secondly, bronchiolitis obliterans is not present in all cases. Moreover, the term COP avoids confusion with airway diseases (constrictive bronchiolitis obliterans).4
Clinical features. Onset of the disease is subacute with progressive cough and dyspnea, often associated with fever (which gives rise to its confusion with respiratory infections), asthenia, and moderate weight loss. In some cases, onset is acute with severe respiratory insufficiency. COP can be idiopathic or associated with collagenosis, infections, medication, or radiotherapy. Chest radio-graphy reveals images of unilateral or bilateral areas of consolidation, occasionally migratory and recurrent. Some cases present with a nodular or small reticulo-nodular pattern. HRCT findings (areas of airspace consolidation with a subpleural or peribronchial distribution) are a useful guide to diagnosis. BAL reveals marked lymphocytosis, often associated with moderate neutrophilia and/or eosinophilia, together with a decreased CD4+/CD8+ T lymphocyte ratio.4
Diagnosis. Diagnosis of COP requires a consistent clinical and radiological picture and histological demonstration of organizing pneumonia in transbronchial or surgical biopsy samples. In this context, BAL abnormalities can help confirm the diagnosis.4
Prognosis. The prognosis is favorable, since the patient usually improves upon treatment with corticosteroids. However, some cases may progress to fibrosis.
Treatment. Oral corticosteroids at the dosage regimen used for NSIP. A recurrence of the disease occurs in 50% to 60% of patients, typically 6 to 12 months after start of treatment when the dose of corticosteroids has usually been reduced to 10 mg or less. In such cases, the dose should be increased to between 20 mg and 30 mg/day. Relapse does not usually modify the long-term prognosis.30 If the patient does not respond to treatment or requires prolonged treatment with corticosteroids, azathioprine at the same dosage regimen used in IPF can be added. However, there is no experience with the effect of this drug in COP.
Acute Interstitial Pneumonia
This is a clinicopathological entity characterized by the presence of diffuse alveolar damage in the lung parenchyma. Diffuse alveolar damage is the histo-pathologic pattern characteristic of acute respiratory distress syndrome. It can be caused by infections, inhalation of toxic products, medication, radiotherapy, and collagen disease. The term AIP should be used exclusively for cases of idiopathic acute respiratory distress syndrome. It is probable that the cases described as Hamman-Rich disease were in reality AIP. The term AIP should not be used in cases of diffuse alveolar damage associated with exacerbations of IPF.4,31
Clinical features. Onset of the disease is acute and is occasionally associated with a flu-like syndrome that progresses to acute respiratory failure requiring mechanical ventilation. Chest radiography reveals bilateral alveolar infiltrates. HRCT evidences ground glass patterns and areas of consolidation, which have been related to the exudative phase of diffuse alveolar damage. In the proliferative and fibrotic phases, traction bronchiectasis and honeycombing become apparent.32
Prognosis. The prognosis in cases of AIP is bad. Patient survival is 50% two months after diagnosis. It has been postulated that cases in the exudative phase of the disease have a better prognosis. Patients who survive may present complete cure, recurrence, or develop a chronic DILD.31,33
Diagnosis. This should be established by surgical lung biopsy.
Treatment. Although no controlled trials have been carried out, treatment with high doses of corticosteroids (100-250 mg/day of intravenous methylprednisolone) has been shown to be effective in the exudative stage of the disease.31
Respiratory Bronchiolitis-Associated Interstitial Lung Disease
RB-ILD is a very rare clinicopathological entity characterized, as its name suggests, by respiratory bronchiolitis associated with DILD. Respiratory bronchiolitis, a lesion caused by smoking, is characterized by pigmented macrophage accumulation in the bronchioles. In some cases, the lesions spread to the alveoli, causing RB-ILD. The disease affects smokers with exposures of 30 pack-years or more. RB-ILD may represent an initial stage of DIP (see "Desquamative Interstitial Pneumonia").4
Clinical features. The clinical signs are those of DILD, although the symptoms are not very obvious. Chest radiographs and HRCT scans show thickening of the walls of the bronchi and ground glass or small reticulonodular patterns. BAL fluid contains hyperpigmented macrophages.
Diagnosis. This should be established by surgical lung biopsy.
Prognosis. The disease evolves favorably, and there are no after effects.
Treatment. Stopping smoking or reducing the number of cigarettes smoked per day cures the disease. If symptoms and/or radiographic or lung function abnormalities persist, corticosteroids should be administered at the dosage regimen used in NSIP.4
Desquamative Interstitial Pneumonia
This is an entity characterized by intra-alveolar macrophage accumulation. It is considered to represent a more advanced stage of RB-ILD because of its similar pathology and association with cigarette smoking. Until a few years ago, DIP was considered to represent the inflammatory stage of IPF, but it has been demonstrated that there is no relationship between the two diseases.4,23
Clinical features. Onset of the disease is insidious or subacute and is characterized by cough and exertional dyspnea, but no constitutional symptoms. In some cases, the disease may progress to severe respiratory insufficiency. Clubbing occurs in 50% of patients. DIP is rarely found in association with other diseases. Cases associated with collagen diseases have been described, although, since all these cases involved smokers, it is possible that tobacco and not collagenosis was the cause of the DIP. Chest radiograph and HRCT scans are not diagnostic and reveal diffuse ground glass opacities sometimes associated with reticular lines. As in the case of RB-ILD, BAL fluid contains hyperpigmented macrophages with variable abnormalities in their cellular form.
Diagnosis. This is established using surgical lung biopsy.
Prognosis. The prognosis is good, with improvement or cure being achieved by tobacco cessation and treatment with corticosteroids.4
Treatment. Patients must stop smoking and are treated with corticosteroids at the dosage regimen recommended for NSIP.
Lymphoid Interstitial Pneumonia (LIP)
LIP is an entity characterized by interstitial lymphoid infiltrates in the lung parenchyma. It was previously considered to be a pulmonary lympho-proliferative disorder, a precursor to pulmonary lymphoma. However, it has now been demonstrated that LIP is only associated with lymphomas in rare cases.4,23
Clinical features. Onset of the disease is subacute, with cough, exertional dyspnea, and associated constitutional symptoms (arthralgias, fever, and weight loss). Blood tests often reveal anemia and hypergammaglobulinemia. LIP is usually associated with other disorders, such as collagen diseases, autoimmune disorders (Hashimoto's disease, myasthenia gravis, pernicious anemia, primary biliary cirrhosis), and immunodeficiencies (agamma-globulinemia). Chest radiograph and HRCT are not diagnostic and reveal ground glass opacities, although reticular and nodular patterns may also occur. BAL is not diagnostic and reveals lymphocytosis.
Diagnosis. Diagnosis is established by surgical lung biopsy.
Prognosis. Seventy percent of patients respond to treatment with corticosteroids. In 20% to 30% of patients, the disease progresses to pulmonary fibrosis.4
Treatment. Administration of corticosteroids at the dosage regimens recommended for NSIP.
Sarcoidosis
Sarcoidosis is a systemic granulomatous disease of unknown etiology which usually affects the lung and thoracic lymph nodes, more rarely the skin and eyes, and occasionally other organs.34,35 Sarcoidosis is probably the result of exposure to environmental factors that provoke an immunological response in genetically predisposed individuals. Its incidence is difficult to estimate since the disease is often asymptomatic and dependent on a geographical area and ethnic group. Incidence in Spain is estimated to be around 1.36 cases per 100 000 inhabitants.36
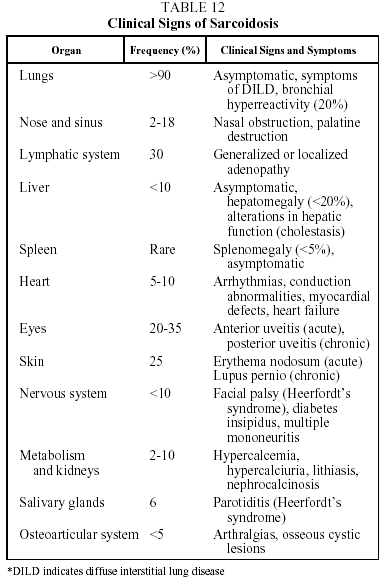
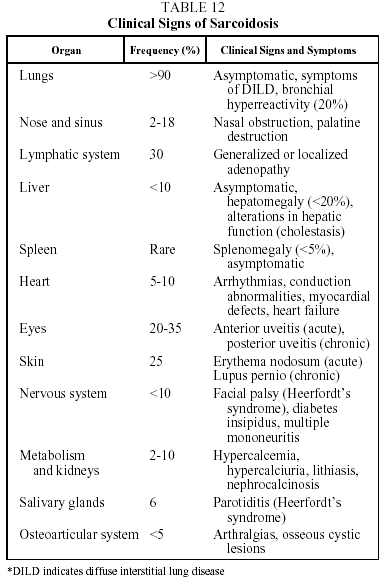
Clinical features. This disease is found mainly in adults under 40 years of age. Presentation is highly variable and can be acute, subacute or chronic. Patients may be asymptomatic (30% of cases) or present with clinical signs that are systemic and/or related to the affected organ. The prototype of acute sarcoidosis is Löfgren's syndrome, which takes the form of fever, arthralgias (mainly in the ankles), erythema nodosum, and symmetrical bilateral hilar lymphadenopathy with or without right paratracheal adenopathy. Another, less common, form of acute presentation is Heerfordt syndrome (anterior uveitis, parotiditis, facial paralysis and fever). Chronic sarcoidosis, often recurrent, is characterized by symptoms related to the affected organs, particularly the respiratory organs.34,35 Clinical signs and their frequency are shown in Table 12.
The two most characteristic findings of chest radiography (hilar lymphadenopathy and lung infiltrates) serve as a basis for staging the disease (Table 13).37

HRCT scans are a useful tool in establishing a diagnosis because they confirm the presence of enlarged nodes and pulmonary infiltrates. The characteristic findings are fine nodular and reticulonodular patterns distributed primarily in the peribronchovascular and subpleural regions. BAL reveals lymphocytosis and an increase in the CD4+/CD8+ T lymphocyte ratio. A CD4+/CD8+ ratio greater than 3.5 is highly characteristic of sarcoidosis and has a specificity of 94%.37 Gallium-67 scans of the lung are of no use in assessing the disease. Whole body gallium imaging may be helpful in a few very specific and difficult-to-diagnose cases.
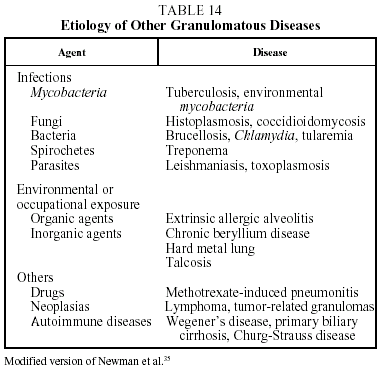
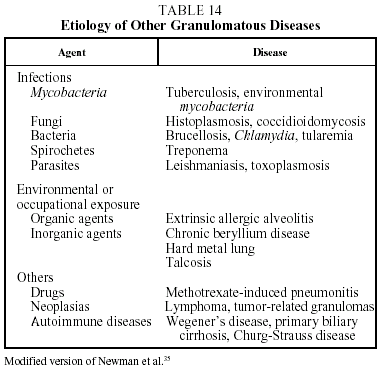
Diagnosis. The diagnosis of sarcoidosis is established by exclusion. It always requires: a) a compatible clinical and radiographic picture; b) the presence of sarcoid granulomas in histologic samples; and c) the exclusion of other diseases capable of producing a similar clinical or histological picture, in particular other granulomatous diseases (Table 14).37 Transbronchial biopsy is the most useful technique for demonstrating granulomas because of its high sensitivity (80%-90%). The decision regarding which other organs should be biopsied will depend on which organs are affected. When a histologic diagnosis cannot be obtained, a high probability diagnosis is acceptable in the following situations: a) Löfgren's syndrome, and b) a compatible clinical and radiological picture together with a CD4/CD8 ratio of 3.5 or greater in BAL.
The Kveim test is no longer used very often because of the difficulty of obtaining the standardized antigen. This is because of poor standardization and because the antigen is a potential transmitter of diseases. The response to delayed cutaneous hypersensitivity tests is reduced during periods when the disease is active, so that these tests could serve as a noninvasive method for monitoring the progress of the disease.8
Prognosis. This is variable since the disease may remit spontaneously or as a result of treatment. In the case of Löfgren's syndrome, spontaneous remission occurs in 85% of cases. The prognosis in pulmonary sarcoidosis is related to the radiographic stage (stage I, remission in 85% of cases within 2 years of diagnosis; stage II, in 40% to 70% of cases; stage III, in 10% to 20% of cases; and stage IV, in 0% of cases). The prognosis is worse for patients over 40 years old, black patients, and patients with symptoms that persist for more than 6 months, or when more than 3 organs are affected or there is concomitant lupus pernio. In extrapulmonary manifestations, prognosis depends on the degree to which the organ is affected and the patient's response to treatment. It is advisable to monitor the evolution of all patients for 3 years after remission of the disease or the end of treatment since recurrence is observed in 10% of cases.37,38
Treatment. The treatment of sarcoidosis is controversial owing to the variability of the initial clinical signs, their severity and progression, and because the disease may remit spontaneously.
Initial treatment takes the form of corticosteroid therapy. There is no consensus on start, duration, dosage, or indications. However, more or less uniform regimens can be drawn up based on the findings of the controlled and open trials that have been carried out.39-41 Its indication is well defined in the case of severe extrapulmonary sarcoidosis, principally in the case of cardiac, neurological, ocular, hepatic, muscular, and cutaneous involvement, and when there is hypercalcemia.
In pulmonary sarcoidosis, corticosteroids are effective in the short to medium term, but no evidence has been adduced to show that they modify the course of the disease.39 Treatment is not indicated in stage I because of the high incidence of spontaneous remission. In stages II and III, treatment is started if the patient presents with symptoms and/or lung functional abnormalities. When no symptoms or functional abnormalities are present, treatment should be started 6 months after diagnosis if interstitial infiltrates persist or when there are signs that the disease is progressing. In stage IV, all patients should be treated.
The initial dose is 40 mg/day of prednisone, or an equivalent dose of another corticosteroid, taken orally for 1 month, after which the dose is gradually reduced (see treatment of NSIP). The recommended duration of treatment is at least 12 months. In stage IV, the initial recommended dose is 1 mg/kg. In the case of relapse, it may be necessary to modify or restart the regimen and maintain an effective dose. Some patients require treatment with a maintenance dose of corticoids for years (<=10-15 mg/day or a double dose on alternate days). If 20 mg/day or more is required, the use of alternative drugs should be considered.42-45
Inhaled corticosteroids have not been shown to be a suitable substitution for the oral presentation. They are, however, indicated in cases with bronchial hyperreactivity and as a maintenance treatment for patients with slight pulmonary sarcoidosis initially treated with oral corticoids.39
In cases of chronic pulmonary sarcoidosis and severe extrapulmonary sarcoidosis, other drugs have been used, such as antimalarial agents (a first line treatment for severe cutaneous and nasal sarcoidosis and hypercalcemia) and immunosuppressive agents, especially methotrexate and azathioprine. However, experience with these therapies is scant.
Immunosuppressive agents have been used alone to treat chronic, corticoid-resistant sarcoidosis, but they are mostly used in combination with corticoids to reduce the dose of the latter. Azathioprine is recommended as a first choice, and methotrexate as a second choice. Other drugs that have been used occasionally are thalidomide and anti-tumor necrosis factor alpha antibodies.42-45 Recommended doses of these drugs are shown in Table 15.

Collagen Diseases
Some patients with collagen diseases (rheumatoid arthritis, systemic sclerosis, dermatomyositis/ polymyositis, systemic lupus erythematosus, Sjögren's syndrome, connective tissue disease) may present DILD during the course of their illness. The DILD associated with collagen diseases are the idiopathic interstitial pneumonias, principally UIP and NSIP46-50 (Table 16).

Clinical features. Clinical, radiographic and lung function manifestations and BAL findings are similar to those of idiopathic interstitial pneumonias without associated collagenosis. Although it is relatively common to find subclinical alveolitis in BAL, the clinical and prognostic significance of this is uncertain. In patients with dermatomyositises/polymyositis, systemic lupus, or sclerosis, concomitant involvement of the respiratory muscles is relatively common. Of particular interest is the antisynthetase or Jo-1 syndrome. This is characterized by the presence of anti-Jo-1 antibodies in patients with polymyositis/dermatomyositis. It is estimated that anti-Jo-1 antibodies are found in 30% of patients with dermatomyositis. The presence of this antibody has been associated with a better response to treatment.51 Patients with systemic sclerosis often present a positive Scl-70 antibody titer.46
Diagnosis. Diagnostic criteria are the same as for idiopathic interstitial pneumonias without associated collagenosis.4,23
Prognosis. Historically it was thought that the prognosis was more favorable for DILD when it was associated with collagen diseases. However, as a result of the new classification of idiopathic interstitial pneumonias, several hospitals have analyzed the histopathological characteristics of cases of DILD associated with collagenosis and have observed that many cases recorded as UIP were in reality NSIP. This could explain the better prognosis in such cases. This is relevant to systemic sclerosis because it has been demonstrated that 80% of the cases of DILD associated with thisase were NSIP.52
Treatment. Treatment is the same as in the case of idiopathic interstitial pneumonias not associated with collagenosis, with the exception of patients suffering from dermatomyositis or systemic sclerosis. In the case of dermatomyositis, the addition of cyclosporin has been shown to be effective in patients whose condition did not respond to corticosteroids and immunosuppressive therapy, mainly when the anti-Jo-1 antibodies were positive.53 In systemic sclerosis with alveolitis (diagnosed on the basis of HRCT findings), the combined administration of cyclophosphamide and corticosteroids has been shown to be effective. The corticosteroids are administered orally (see treatment of NSIP), and cyclophosphamide in bolus form at a dosage of 750 mg/m²/month for 6 months followed by 750 mg/m²/quarterly for 1 year. If improve-ment is observed or alveolitis persists, continuation of treatment for another year is recommended.54
Extrinsic Allergic Alveolitis
EAA, also known as hypersensitivity pneumonitis, is a form of DILD caused by the inhalation of organic agents, although inorganic substances (isocyanates) can also cause this disease. Since the number of organic products that humans may inhale is very considerable, the causes of EAA are increasingly more numerous. The most common forms of EAA are poultry-workers lung and farmer's lung. Other types diagnosed in Spain are aspartosis, mother-of-pearl lung, air conditioner alveolitis, ultrasonic humidifier lung, sausage-cleaners lung, suberosis, isocyanate lung, soy lung, and Candida lung.55-57
Clinical features. Onset of the acute form of this disease usually occurs between 2 and 8 hours after contact with the antigenic source and takes the form of dyspnea, cough, fever, asthenia, presternal chest tightness, joint and muscle pain, chills, and sweating. Chest radiography is characterized by a fine miliary or alveolar pattern, and HRCT reveals patchy ground glass opacities with areas of hyperlucency and a fine nodular pattern. Symptoms disappear spontaneously once contact with the antigen is avoided. The subacute form occurs after continuous, but not massive, inhalations of the causal agent. The clinical picture is characterized by asthenia, weight loss, general malaise, and febricula, in addition to respiratory symptoms. Chest radiography and HRCT finding are similar to those of the acute form. The clinical signs of the chronic form are either similar to those of IPF or else consist of cough and expectoration (a similar clinical picture to that of chronic obstructive pulmonary disease). BAL findings include very marked lymphocytosis, mastocytosis, and a reduced CD4+/CD8+ T lymphocyte ratio, although neutrophiles and CD4 lymphocytes may predominate immediately after exposure. Transbronchial biopsy can reveal the histological changes characteristic of the disease.55,56
Diagnosis. In cases with characteristic clinical signs and a temporary contact with a suspected antigen source, diagnosis can be established with some certainty. Spontaneous improvement upon avoiding contact with the causal antigen, and positive test results to specific IgG (using precipitins or enzyme immunoassay) will help establish diagnosis. Intradermal, immediate-reaction tests using the specific extract and readings 15 min after injection can simulate contact with the antigen and serve as a test that can discriminate between mere contact and the disease.58 Bronchial challenge tests are indicated in difficult-to-diagnose cases and are considered to be the "gold standard" in the diagnosis of EAA. Bronchial challenge testing involves prior preparation of the antigen extract, and lung function tests must evidence FVC and DLCO values above 60% of predicted. The test is considered positive if any of the 3 criteria shown in Table 17 are fulfilled.59

If the test result is negative, it is repeated the following day using a higher concentration. If negative results persist, the test can be repeated for 2 days. The patient can also be exposed directly to the antigen source for 5 days.
In an attempt to standardize the diagnosis of EAA as much as possible, a series of criteria have been proposed (Table 18).60 The diagnosis is confirmed if 4 major criteria and at least 2 of the minor criteria are fulfilled.

Prognosis. When the disease is diagnosed early and contact with the antigen is avoided, prognosis is good, with a complete cure being the outcome in most cases. In 10% to 20% of cases, the disease progresses to pulmonary fibrosis or chronic airway obstruction (chronic forms).
Treatment. This consists of avoiding contact with the antigen. When clinical signs and radiologic or lung function abnormalities persist, corticosteroids are usually administered (see dosage recommended for the treatment of NSIP), although there is no evidence that administration of these drugs modifies the long term prognosis.
Radiotherapy
The lung parenchyma always reacts to radiation on exposure, but symptoms occur in only 5% to 15% of patients. The appearance of DILD depends on several factors: individual susceptibility, the volume of the lung included in the field of radiation, the dose of radiation used, prior history of lung disease, and concomitant therapy with certain cytostatic drugs.61
Clinical features. There are 3 clinical forms of this entity: pneumonitis, fibrosis, and organizing pneumonia. Radiation-induced pneumonitis usually appears 3 weeks to 6 months after exposure, reaching maximum intensity normally at 4 to 6 weeks. It is characterized pathologically by the presence of diffuse alveolar damage. Chest radiography reveals patchy or confluent alveolar patterns, which generally coincide with the irradiate field, but these can extend beyond this area and may even be contralateral. Apart from respiratory symptoms, patients may present febricula and chest pain. Chest radiography and HRCT reveal a reticular or honeycomb pattern, sometimes with traction bronchi-ectasis. Cases of organizing pneumonia after lung radiation have been described, principally in the case of treatment of breast cancer, which can affect the lung contralateral to the irradiation.61
Diagnosis. The diagnosis of radiation-induced pneumonitis and fibrosis is established by a prior history of radiotherapy and a clinical and radiographic picture consistent with this diagnosis. The diagnostic criteria of radiation-induced organizing pneumonia are the same of those of the idiopathic form.
Prognosis. In the case of pneumonitis with a good response to treatment, the prognosis is favorable. The prognosis for fibrosis is variable. Although most patients are only slightly or moderately affected, this condition can occasionally progress to respiratory failure. The prognosis for organizing pneumonia is the same as for idiopathic COP.
Treatment. Corticosteroids at the dosage used for NSIP are indicated for pneumonitis (see treatment of NSIP). However, corticoid therapy has not been shown to be effective in the treatment of fibrosis. Treatment of radiation-induced organizing pneumonia is the same as for idiopathic COP.61
Drugs
The drugs that have the potential to cause DILD are numerous. They include chemotherapy agents, antiarrhythmics, antibiotics, anticonvulsants, antiinflammatory agents, and illegal drugs. The elaboration of an exhaustive list of such drugs is not the object of these guidelines, so we refer interested readers to the Internet URL http://www.pneumotox.com. Most of the literature deals with isolated clinical cases or short series of cases, so that the epidemiology is not well defined. Moreover, there are no well-defined criteria that establish whether a given clinicopathological picture is caused by a drug. With most drugs, no factors predisposing patients to their lung toxicity have been identified, making such reactions idiosyncratic. With others, factors favoring lung toxicity include a higher accumulated dose, the type of disease being treated, and concurrent treatment with chemotherapy agents, radiotherapy, or high concentrations of oxygen.62
Clinical features. Drugs can cause various forms of DILD: UIP, diffuse alveolar damage, organizing pneumonia, LIP, DIP, NSIP, pulmonary eosinophilia, and acute hypersensitivity pneumonitis. The clinical and radiologic signs of each form of drug-induced DILD are similar to those of the idiopathic form of the same disease. BAL results reveal variable cell profiles, and the CD4+/CD8+ T lymphocyte ratio is often reduced.
Diagnosis. Diagnosis is established by a prior history of drug use in conjunction with a clinical and radiographic picture consistent with DILD. Drug-induced DILD may also be suspected because the clinical signs have disappeared when treatment with a particular drug is discontinued and reappeared on resumption of treatment.
Prognosis. This depends on the form of DILD caused by the drug.
Treatment. Interruption of treatment with the suspected drug. When the clinical, radiological, and/or lung function abnormalities persist, treatment with corticosteroids is indicated (see treatment of NSIP).
Lymphangioleiomyomatosis
LAM is an uncommon multisystemic disease of unknown cause. It is found almost exclusively in women of childbearing age, making it probable that hormonal factors intervene in its pathogenesis. LAM is characterized by the abnormal proliferation of smooth muscle cells and may affect, in addition to the lungs (smooth peribronchiolar, perivascular, and perilymphatic muscle), other organs, such as the kidneys, peritoneal lymphatics, liver, uterus, and pancreas.63-66
Clinical features. Apart from the symptoms typical of DILD, other signs of this disease are: recurrent pneumothorax (69%), chylothorax (23%), hemoptysis (20%), and less often, ascites, pericardial pleural effusion, chyloptysis, and chyluria. There is a frequent association (60% of cases) with renal angiolipomas. In 25% of cases, LAM is associated with tuberous sclerosis, a hereditary disease characterized by multiorgan hamartomatosis.67,68 In the early stages of the disease, chest radiography and HRCT reveal micronodular pseudo-miliary opacities and B Kerley lines. In more advanced stages, thin-walled cyst images appear, predominantly in the basal zones. In an appropriate clinical context, HRCT images are highly indicative of the diagnosis.
Diagnosis. This is established by transbronchial or surgical lung biopsy. The HMB-45 monoclonal anti-body selectively stains the muscular proliferation of LAM, even in transbronchial biopsy samples.63
Prognosis. Most cases progress to diffuse microcystic destruction of the lungs, which leads to severe respiratory failure. In some cases improvement or stabilization has been observed with hormonal treatment.
Treatment. Various kinds of hormonal treatment have been used with variable results, but in no case have these led to a cure. The best results have been obtained with a regimen of intramuscular medroxyprogesterone acetate at a dosage of 400-800 mg/month for 1 year. If no improvement is observed, bilateral ovariectomy is recommended. Treatment with tamoxifen is not advisable because this medication has been associated with worsening of the disease.63
Histiocytosis X
Langerhans cell histiocytosis is a multisystemic pathological entity with diverse clinical profiles depending on the age of the patient and the extension of the area affected by the Langerhans cell proliferation characteristic of the disease. Langerhans cell histiocytosis is a group of disorders that includes an acute form of the disorder which affects younger infants (Letterer-Siwe disease), a multifocal eosinophilic granuloma which affects slightly older children (Hand-Schüller-Christian disease), and histiocytosis X (also called eosinophilic granuloma or granulomatosis of Langerhans cells) which, in the adult form, is usually located in the lung. Smoking has been implicated in the pathogenesis of histocytosis X because the disease is rare in nonsmokers, and the lungs of smokers contain more Langerhans cells than those of nonsmokers.69,70
Clinical features. The disease affects young adults smokers, and presents in the form of cough and progressive exertional dyspnea. Half of the patients are asymptomatic in the early stages. Pneumothorax is common (25% of cases) and can be the first symptom. Associated signs may include generally asymptomatic diabetes insipidus (28% of cases) and cystic bone lesions in the cranium, long bones, ribs, and pelvis. Chest radiography reveals interstitial infiltrates with small air cysts in a predominantly apical distribution. HRCT clearly reveals the air-filled cysts with well-defined walls, and its findings help establish diagnosis.69-71 Recent studies have shown that somatostatin receptor scintigraphy can be a highly sensitive technique for detecting disease activity and monitoring response to treatment.
Diagnosis. This is established using HRCT findings, transbronchial biopsy and BAL (CD1+ cells >5% of cells of macrophage lineage). If there is any doubt, a surgical lung biopsy should be performed. The presence of Birbeck granules visible under electron microscope in the BAL and transbronchial biopsy is a diagnostic sign. The S-100 antibody has also been used on lung biopsy samples to diagnose this condition.69,70
Prognosis. The prognosis is variable since the disease may remit spontaneously, remain stable, or progress to pulmonary fibrosis. The functional and radiographic abnormalities and the symptoms may improve if the patient stops smoking.69,70,73
Treatment. This consists in stopping smoking. Corticosteroids at the dosage recommended for other DILD (see treatment of NSIP) can be effective in the initial stages of the disease. No controlled trials have investigated the efficacy of any other drugs.70,72
Alveolar Proteinosis
This is a rare clinical entity. Its incidence is estimated at 1 case per 2 000 000 inhabitants. It is characterized by deposition in the alveoli of components of the surfactant. Three clinical forms have been described: congenital, primary or idiopathic, and secondary. The pathogenesis is unknown, although recent studies have pointed to abnormalities in the homeostasis of the surfactant as a result of inhibition of the action of granulocyte-macrophage colony-stimulator factor (GM-CSF) in the primary form of the disease, and genetic mutations in the congenital form.74
Clinical features. The disease is normally diagnosed in patients between 20 and 50 years old, although cases have been reported in neonates and elderly patients. Primary alveolar proteinosis is the most common form (90% of cases). Secondary alveolar proteinosis is associated with certain hematological disorders (myeloid leukemia, lymphoma, Fanconi's anemia, IgG monoclonal gammopathy), exposure to inorganic dusts (silicate, aluminium, and titanium), drugs (busulfan, chlorambucil), and infection with the human immunodeficiency virus. The association with infections (Nocardia asteroides, Mycobacterium tuberculosis and other opportunistic pathogens) may be a cause or a consequence of the disease.
The disease usually presents as cough and exertional dyspnea. Some 30% of patients are asymptomatic. Fever indicates the presence of a superinfection, although prolonged low-grade fever can be an expression of the disease itself. Patients present with digital clubbing in 30% of cases. Serum lactate dehydrogenase levels are often elevated. By plain chest radiograph, bilateral and symmetrical alveolar infiltrates may be seen, more marked in the perihilar zones (butterfly wings), but these may be predominantly peripheral or basal and, in some cases, are the only finding in asymptomatic patients. HRCT may reveal ground glass opacities, consolidation of the air space with thickening of the interlobular septa (resulting in a "crazy-paving" pattern).74
Diagnosis. Diagnosis can be established on the basis of BAL and transbronchial biopsy findings. Fluid obtained from BAL has a milky appearance. Analysis reveals material that is PAS positive and Alcian blue negative, a high concentration of total phospholipids (phosphatidylglycerol), and the presence of lamellar bodies visible on electron microscopic examination. Detection of serum autoantibodies against GM-CSF is useful in the serological diagnosis of idiopathic alveolar proteinosis owing to its high sensitivity and specificity.74
Prognosis. This is variable. The disease remits spontaneously in 25% of cases, although it can progress to respiratory failure. The chief factors that define the course of the disease are pulmonary infections caused by opportunistic pathogens.74
Treatment. This consists of serial therapeutic BAL, which are indicated in cases of progressive disease.75,76 Recently, some authors have reported on cases treated with GM-CSF with good results.77
Alveolar Microlithiasis
Alveolar microlithiasis is a rare disease of unknown etiology, characterized by the presence of calcified bodies (microliths) inside the alveolar spaces. The fact that only around 300 cases are described in the literature is an indication of the very low prevalence of this disorder. Around 60% of cases present a marked family relationship, especially in monozygotic twins, which suggests recessive autosomal transmission.78,79
Clinical features. This disease is more common in the third, fourth, and fifth decades of life, although cases have been reported in infants and the elderly. Some 70% of patients are asymptomatic at the time of diagnosis. Chest radiography and HRCT findings are characteristic and reveal a diffuse bilateral pattern of micronodules (1 mm-5 mm). Obliteration of the heart borders and diaphragm in conjunction with an increase in linear radiolucency between the parenchyma and the rib cage (the so-called black pleural line) is considered characteristic of this disease.80
Diagnosis. This is established on the basis of the radiographic findings and those of transbronchial or surgical lung biopsy.79
Prognosis. The course of the disease is variable. As many cases have been described with a course of more than 30 years without clinical deterioration as there have been cases that progressed to pulmonary fibrosis and respiratory failure.78,79
Treatment. There is no specific treatment for this disease.
Pulmonary Eosinophilias
This is a clinical syndrome encompassing diverse processes that give rise to peripheral eosinophilia and pulmonary infiltrates with eosinophilia.81 Table 19 shows the causes of pulmonary eosinophilia. The most common cause is chronic eosinophilic pneumonia.

The parasites most often associated with pulmonary eosinophilia are Toxocara and Ascaris. Certain drugs cause pulmonary eosinophilia, and information on these can be found on the Internet URL www.pneumotox.com. The effects these drugs have on the lungs are periodically updated on this web site. In addition, certain clinical entities can be associated with pulmonary eosinophilia, although this is rare and is not one of the predominate characteristics of the clinical picture. These entities include some infectious diseases (histoplasmosis, coccidioidomycosis) and neoplasias (Hodgkin's disease).
Clinical features. The clinical picture of pulmonary eosinophilias is characterized by symptoms typical of DILD--fever, asthenia, weight loss, and night sweats--which explains why it is sometimes confused with pulmonary infections. The characteristic findings of chest radiography are changing and recurrent peripheral alveolar infiltrates distributed predominantly in the upper lung fields. HRCT can aid diagnosis in cases with atypical chest radiography results. Peripheral eosino-philia is present in 85% of cases. When the disease is active, eosinophilia is always present in BAL, generally representing over 40% of total cells. Acute eosinophilic pneumonia is an acute feverish disease of short duration characterized by respiratory insufficiency which generally requires mechanical ventilation. Chest radiography shows bilateral alveolar infiltrates.81
Diagnosis. Diagnosis is established by means of a compatible clinical and radiographic picture, eosinophilia in peripheral blood and BAL, or by transbronchial biopsy. Allergic bronchopulmonary aspergillosis, drug-related disease, vasculitis, and parasitosis should always be excluded. The diagnostic criteria for allergic bronchopulmonary aspergillosis are different in the United Kingdom and the USA. The criteria commonly used are shown in Table 20, together with those of Rosenberg et al.82,83 Rosenberg et al83 include positive precipitin test results against Aspergillus, proximal bronchiectasis, and elevated serum IgE among the major criteria. In order to rule out the possibility of parasite infestation, 3 measurements of parasites in feces should be performed, 1 every 15 days, in order to cover the passage through the intestine during the cycle of the parasite.

Prognosis. This depends on the etiology. Pulmonary eosinophilias caused by drugs and helminths progress towards a cure once the causal agent has been eliminated. Recurrence of the clinical signs and symptoms is common in the case of bronchopulmonary aspergillosis, and in some cases the disease causes irreversible airway obstruction. In 50% of cases, patients with chronic eosinophile pneumonia suffer relapse when corticosteroid treatment is stopped. The prognosis for acute eosinophilic pneumonia is favorable and recurrence is uncommon. Pulmonary infiltrates generally disappear within 48 to 72 hours of administration of corticosteroids.81,84
Treatment. Oral corticosteroids at a dosage of 0.5-1 mg/kg per day. The dose is gradually reduced depending on the course of the disease. Treatment should generally be maintained for 6 to 12 months, and a low dose is occasionally required for several years. The treatment for acute eosinophilic pneumonia is high doses of methylprednisolone (1-2 mg/kg every 6 hours for 2 to 3 days), followed by a lower dose (0.5 mg/kg for 2 weeks) and gradual tapering of the dose until treatment is completed.81,84
Pneumoconiosis
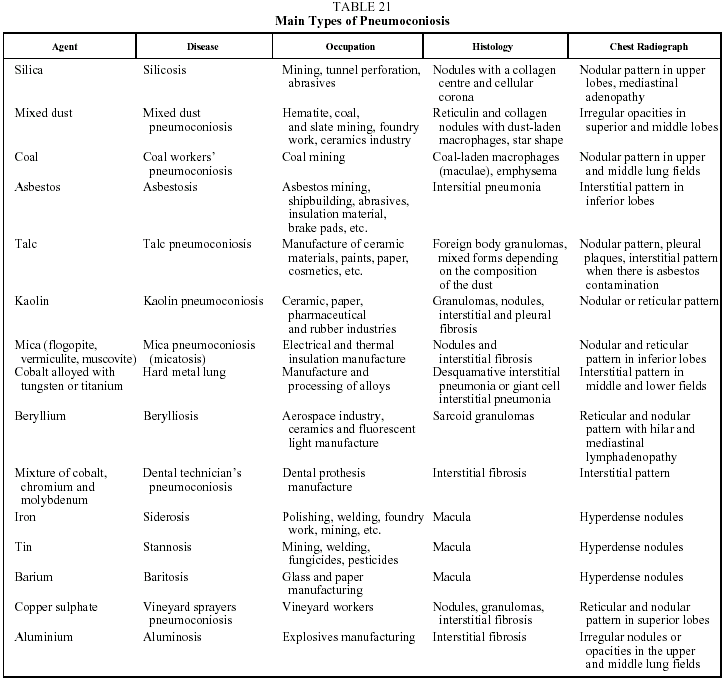
This is a DILD caused by the inhalation of inorganic dust particles. A common characteristic of all forms of the disease is a period of latency--usually more than 20 years--between exposure and the onset of symptoms or diagnosis. It is also interesting to note that there is a direct relationship between the degree of exposure (including both intensity and duration) and the quantity of dust deposited in the lung. Table 21 lists the inorganic agents that most often cause pneumoconiosis on inhalation.85-87
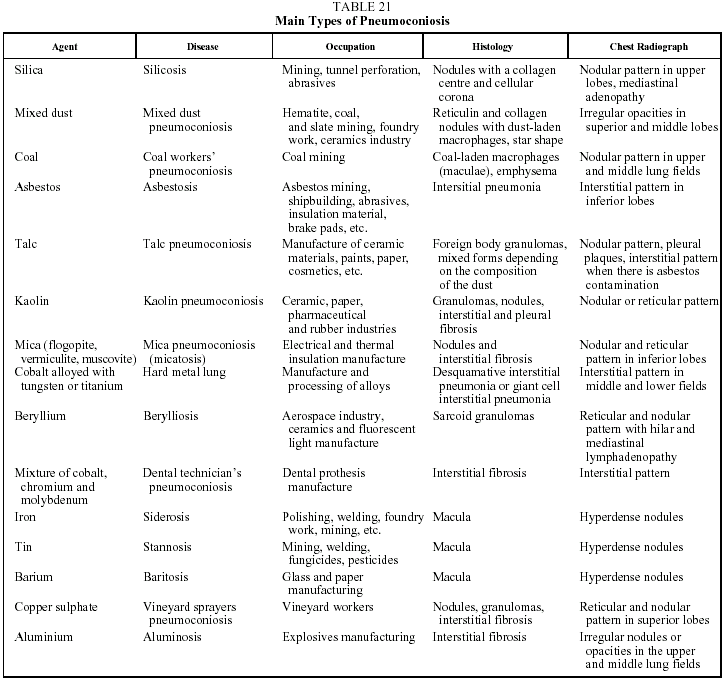
The findings of lung histology vary from the formation of silicotic nodules in the case of silicosis, to interstitial fibrosis in the case of asbestos, to the simple deposition of iron particles in the lung in siderosis. In many cases, the exposed individual inhales dust containing silicon dioxide and silicates in variable proportions or dust contaminated with asbestos fibers, which explains the variability of the histologic findings and of the clinical and radiological manifestations of the mixed forms of the disease.
Clinical features. The clinical picture is the same as that of other forms of DILD. Chest radiography is the recommended technique for detecting the presence of pneumonoconiosis. It has been used as a screening method in on-the-job checkups, especially in the case of miners. An agreement exists on the reading and classification of radiographic abnormalities in the lung.
The International Labour Office (ILO) published its most recent guidelines on reading chest images in 1980. These cover the detection of nodular and interstitial lung lesions, and an appendix on the subject of pleural lesions. The radiologists and pneumologists who read radiographs and classify them according to the ILO guidelines require prior training. However, since the advent of HRCT, this classification system has been used less often.87
Chest HRCT affords a better definition of pleural lesions and of pulmonary pneumoconiotic lesions under 2 mm. It is the recommended procedure when chest radiography findings are not diagnostic, or when associated diseases, such as tuberculosis, pulmonary neoplasia, or emphysema, are suspected.
Diagnosis. This is based on the existence of a clinical picture and radiographic results consistent with this diagnosis in conjunction with a prior history of exposure to potentially causal inorganic agents.
In a few cases, medical history, clinical signs, and radiographic results are not sufficient to establish a conclusive diagnosis. In such cases, histological examination of the lung may be necessary. A mineralogical examination of lung samples is occasionally necessary to demonstrate that a pulmonary deposit of the inorganic agent exists. It is advisable to perform this analysis on surgical samples, since transbronchial biopsy samples are not representative of the lung as a whole. Since a certain quantity of inorganic lung deposit exists in the general population, the mineralogical analysis should be quantified and compared to the predicted values for the relevant population. Analysis using optical and polarized light microscopy allows the detection of birefringent elements, such as asbestos bodies, nonfibrous silicates, and silica. However, most particles or fibers smaller than 5 µ are not visible, and it is not possible to determine their nature by chemical analysis. Quantifying the inorganic dust deposit in the lung requires the destruction of the organic material by incineration or chemical digestion and the identification of fiber particles using electron microscopy and analytic methods (techniques outside the scope of most hospitals specializing in respiratory medicine in Spain). The most accessible technique is the energy dispersion analysis of x-rays (EDAX). The result is expressed as the number of inorganic elements per gram of dry lung. In the case of asbestos, exposure can be confirmed using BAL.87 It is considered that the presence of 1 or more asbestos bodies per milliliter indicates a workplace exposure to this agent.11
Treatment. There is no effective treatment once the disease has begun. An exception is the case of berylliosis, which can improve upon treatment with corticosteroids.
Other Diffuse Interstitial Lung Diseases
Inflammatory diseases of the intestine. Ulcerous colitis and Crohn's disease are, in very rare cases, associated with certain DILD (COP, NSIP, UIP, DIP, pulmonary eosinophilia). Sometimes these DILD are caused by the medication that the patient is taking (sulfasalazine or melphalan). Whipple's disease can be associated with DILD and the radiographic characteristics are similar to those of sarcoidosis (hilar lymphadenopathy and intersitial pattern).88
Amyloidosis. Amyloidosis may cause a form of DILD with no special characteristics. The prognosis is bad since the disease progresses within only a few years and there is no effective treatment.1
Acknowledgments
Dr. J. Ferrer (Hospital Vall d'Hebron, Barcelona) collaborated in the section on pneumoconiosis. Dr. C. Almonacid (Hospital de La Princesa, Madrid) collaborated on the section on diagnostic methods. Dr. J. Gaudó (Hospital Ramón y Cajal, Madrid) collaborated on the section on lymphangioleiomyomatosis. Dr J. Font and Dr R. Cervera (Servicio de Enfermedades Autoinmunes y Sistémicas, Hospital Clínic, Barcelona) collaborated on the therapeutic regimens for DILD associated with systemic sclerosis.
Correspondence: Dr. A. Xaubet.
Servei de Pneumologia. Hospital Clínic.
Villarroel, 170. 08036 Barcelona. España.
E-mail: axaubet@clinic.ub.es
Manuscript received March 18, 2003. Accepted for publication March 18, 2003.







