





Pleuroparenchymal fibroelastosis (PPFE) is a rare disease that has been recently included in the updated consensus on idiopathic interstitial pneumonias. It shares some clinical features with other chronic interstitial pneumonias (dyspnea, dry cough), and is radiologically characterized by pleural and subpleural parenchymal fibrosis and elastosis, mainly in the upper lobes. The main histological findings include pleural fibrosis and prominent subpleural and parenchymal fibroelastosis. Its characterization is based on the increasing number of cases reported in the literature, so several aspects of the etiology, pathogenesis and natural history are still unknown. Although some cases have been described as idiopathic, PPFE has been reported as a complication after bone marrow transplantation, lung transplantation and chemotherapy, especially with alkylating agents.Spontaneous or iatrogenic pneumothorax is a frequently reported complication of invasive diagnostic tests for identifying PPFE. The disease course is variable, ranging from slow progression to rapid clinical deterioration. No treatment has shown evidence of efficacy, and lung transplantation remains the only option for patients who fulfill the diagnostic criteria for this option. Recognizing and disseminating the specific features of PPFE are essential to raise the level of clinical suspicion for this entity, and to implement appropiate diagnostic and management by the multidisciplinary team.
La fibroelastosis pleuropulmonar (FEPP) es una entidad rara que ha sido recientemente incluida en el último consenso internacional multidisciplinar sobre neumonías intersticiales idiopáticas. Comparte rasgos clínicos con otras neumonías intersticiales crónicas (disnea de esfuerzo, tos seca, entre otros), y radiológicamente se caracteriza por afectación pleural y pulmonar de predominio en lóbulos superiores. Sus principales hallazgos histológicos incluyen fibrosis de la pleura visceral con prominente fibroelastosis subpleural y parenquimatosa. El conocimiento de sus características se basa en el creciente número de casos descritos en la literatura, por lo cual se desconocen varios aspectos sobre su etiología, patogénesis e historia natural. Aunque algunos casos han sido identificados como idiopáticos, la FEPP ha sido asociada como complicación tras el trasplante de médula ósea, trasplante pulmonar y tratamiento con quimioterapia, especialmente con agentes alquilantes. El neumotórax espontáneo o iatrogénico tras las pruebas invasivas para su diagnóstico es una complicación frecuentemente comunicada. La evolución descrita de la FEPP es variable, desde lentamente progresiva hasta casos con rápido deterioro clínico. No hay ningún tratamiento con evidencia de eficacia, y el trasplante pulmonar se erige como la única opción en aquellos pacientes que cumplan los criterios. El reconocimiento y difusión de las características de esta entidad son fundamentales para elevar el grado de sospecha clínica y permitir un adecuado manejo diagnóstico por parte del equipo multidisciplinar.
Pleuroparenchymal fibroelastosis (PPFE) is a clinical–pathological entity included in the group of rare idiopathic interstitial pneumonias (IIPs) in the latest classification published by the American Thoracic Society/European Respiratory Society.1 Some case series published in the early 1990s, particularly in non-English language journals, describe pulmonary upper lobe fibrosis (Amitani's disease) with characteristics similar to PPFE.2,3 However, the entity was first officially characterized in 2004 by Frankel et al.4 in a description of radiological and histological clinical findings from a series of 5 patients. The group called for PPFE to be classified as a specific entity within the group of interstitial lung diseases (ILD). Since then, around 100 cases of PPFE have been published, adding to the understanding of the characteristics of this particular entity.5–24 PPFE is characterized by predominantly upper lobe pleural and parenchymal involvement, with pleural fibrosis and subpleural fibroelastosis. Pneumothorax is a common complication in these patients, and is at times the primary manifestation of the disease.2,8,21 The etiology of PPFE is unclear, although the identification of the disease in bone marrow and lung transplant recipients and in patients who have undergone chemotherapy is generating increasing interest among researchers. PPFE has also been associated with recurrent lung infections and immune disorders, and cases of a familial form of the disease have also been reported.7,11
In this review, we will discuss the most relevant aspects of PPFE, with particular emphasis on the different diagnostic criteria put forward. We will also comment briefly on new pathophysiological hypotheses that the entity could be the final response to different insults to the lung, suggesting that it could constitute a new pathological phenotype within a spectrum of disorders, such as chronic pulmonary allograft dysfunction.
Epidemiological Characteristics and Associated EntitiesAccording to studies published so far, age at onset of PPFE varies widely, with a mean of 57 years,25,26 although it most commonly occurs in young adults aged between 20 and 30 years. There is no clear gender predominance.
Smoking habit does not seem to be a risk factor for this disease. Some series, however, have reported prior exposure to asbestos, aluminum and avian antigens.2,7,11
Prior cancer treatment with radiotherapy and chemotherapy is often described in association with PPFE, particularly when alkylating agents such as cyclophosphamide or nitrosoureas are used.20,27,28 The time from administration of these agents and development of PPFE varies greatly in the studies published, ranging from 6 months to 16 years.28
Several studies have described PPFE as a complication of bone marrow and lung transplantation.8,10,11,13,16,23,24 In some of these cases, authors have reported the presence of concurrent foci of bronchiolitis obliterans (BO) or diffuse alveolar damage (DAD). Some authors have suggested that DAD could precede PPFE.10,24,26 Others have speculated that PPFE could be a restrictive phenotype, functionally and histologically different from the classic BO syndrome, within the spectrum of manifestations of chronic lung allograft dysfunction. BO is characterized by an irreversible decline in forced expiratory volume in 1s (FEV1) and fibrotic obliteration of airway lumens in transplanted lungs. When organ rejection is accompanied by the histological changes typical of PPFE, decline in FEV1 is joined by a decrease in forced vital capacity (FVC) and total lung capacity (TLC), and prognosis is poorer than in BO syndrome.10,24,26
This association also suggests the existence of a possible autoimmune element in its pathogenesis. Some authors have reported elevated levels of antinuclear antibodies and rheumatoid factor,4,11 together with concurrent autoimmune diseases such as ankylosing spondylitis or ulcerative colitis, among others.3
Both Frankel et al.4 and Reddy et al.11 have reported patients with a family history of ILD. In some cases, a history of recurrent respiratory infections prior to PPFE diagnosis has led to suggestions that repeated inflammation could be a risk factor for the disease.11–13Table 1 summarizes the different entities associated with PPFE.
Entities Associated with PPFE.
| Idiopathic PPFE |
| Bone marrow transplantation |
| Chemotherapy |
| Lung transplantation |
| Radiotherapy |
| Respiratory infections: recurrent bronchitis, Aspergillus, MAI |
| Autoimmune diseases: AS, ulcerative colitis, psoriasis, RA |
| Workplace exposure: asbestos, aluminum |
| Hypersensitivity pneumonitis |
| Familial PPFE |
AS: ankylosing spondylitis; MAI: intracellular Mycobacterium avium; PPFE: pleuroparenchymal fibroelastosis; RA: rheumatoid arthritis.
Adapted from: Watanabe.3
The most common presentation is dyspnea on exertion and insidious onset dry cough. Weight loss is also a common symptom.
Pleuritic chest pain, possibly secondary to spontaneous pneumothorax, can also be the presenting symptom in some patients. Pneumothorax, a characteristic complication in the natural history of PPFE, can be secondary to parenchymal changes such as cysts in areas of apical fibrosis, altered resistance of the pleura to shear stress, or hypovascularization of the affected area.26,28 Spontaneous pneumothorax resorption is uncommon in PPFE, while persistent leaks following treatment and slow re-expansion of the affected lung are commonplace.8,21,28 Iatrogenic pneumothorax can also occur following surgical biopsy.
Finger clubbing and dry “velcro” crackles are far rarer in PPFE than in other IIPs such as idiopathic pulmonary fibrosis (IPF).3
Chest deformity, or flattened thoracic cage (caused by a reduced anterior–posterior diameter in relation to the transverse diameter of the chest) is a common clinical sign and an indication of disease progression.3,28–30 Several Asian series have been published describing patients with body mass index in the low or lower limit of normal range (slender stature).3,9,12,29
Functional CharacteristicsThe restrictive breathing pattern is similar to that found in IPF, and presents with an increased FEV1/FVC ratio and a decrease in FVC and TLC. However, an increased residual volume (RV)/TLC ratio is also found. This functional characteristic is caused by upper lobe collapse due to fibrosis, which can lead to compensatory hyperinsuflation in lower lobes.3,9 Diffusing capacity (DLCO) is reduced and DLCO corrected for alveolar volume (DLCO/VA) can be either normal or slightly reduced. Arterial hypoxemia and hypercapnia present as the disease progresses. Watanabe et al.12 described a rapid mean decline in FVC of 20% measured over 1 year in 7 patients. The same rapid decline in FVC was also a significant finding in a recently published study in 12 Asian patients.29 This seems to suggest that PPFE evolution has a greater tendency towards rapid progression.
Radiological CharacteristicsRadiological findings can be an essential element in the diagnosis of suspected PPFE. The most common finding in chest radiography is irregular pleural thickening in bilateral apical areas of the lungs in the initial stages of the disease. This can be an incidental finding in asymptomatic patients,3 and is easily confused with apical caps.
This is followed by upper hilar retraction and diaphragmatic elevation, with loss of volume in upper lobes (Fig. 1a). A characteristic finding in PPFE is “flattened thoracic cage”, consistent with the signs observed during the clinical examination. This finding appears in the lateral view on chest radiographs as a marked decrease in the anterior–posterior diameter of the thorax3 (Fig. 1b). As the disease progresses, subpleural reticular and nodular opacification can be seen in the upper lobes.4 Initially, these abnormalities are confined to the upper lobes. Over the course of the disease, signs of fibrosis with loss of volume, or bullae or cysts in the upper lobes, can appear. Fibrosis can also extend to adjacent lobes.3,11
 Chest radiograph (posterior anterior view), showing evolution of pleuroparenchymal fibroelastosis, characterized by progressive loss of volume in upper lobes, upper bilateral hilar retraction, and biapical pleural thickening. (b) Chest radiograph (lateral view), showing flattened thoracic cage; note how the anterior posterior diameter of the chest (arrow) has narrowed relative to the craniocaudal diameter.")
(a) Chest radiograph (posterior anterior view), showing evolution of pleuroparenchymal fibroelastosis, characterized by progressive loss of volume in upper lobes, upper bilateral hilar retraction, and biapical pleural thickening. (b) Chest radiograph (lateral view), showing flattened thoracic cage; note how the anterior posterior diameter of the chest (arrow) has narrowed relative to the craniocaudal diameter.
Prominent pleural thickening associated with signs of fibrosis (traction bronchiectasis, architectural distortion and loss of volume), and reticulation, notably in the upper lobes, are seen on high resolution computed tomography (HRCT)7,9,11 (Fig. 2). As the disease progresses, the reticular opacities and signs of fibrosis extend to the remaining lobes (Fig. 2b). In contrast to the more pronounced findings in the upper lobes, radiological changes at the lung bases are less obvious or more discreet, and can even be totally absent (Fig. 2c).
, showing evolution of the above case over a period of 2 years and 2 months. The top image (a) shows only a few discrete elongated opacities in both apices; the lower image (b) shows loss of volume and pleuroparenchymal fibrotic infiltrates. (c) Shows an HRCT slice at the level of the tracheal carina. The image shows architectural distortion, with pleural thickening and elongated fibrotic parenchymal opacities (lung window).")
High resolution computed tomography (HRCT), showing evolution of the above case over a period of 2 years and 2 months. The top image (a) shows only a few discrete elongated opacities in both apices; the lower image (b) shows loss of volume and pleuroparenchymal fibrotic infiltrates. (c) Shows an HRCT slice at the level of the tracheal carina. The image shows architectural distortion, with pleural thickening and elongated fibrotic parenchymal opacities (lung window).
In the series of 12 patients described by Reddy et al.,11 HRCT findings showed fibrotic changes distal to areas of pleuroparenchymal thickening, mimicking a usual interstitial pneumonia (UIP) or non-specific interstitial pneumonia pattern. Moreover, of the 6 patients with follow-up radiographs, 5 showed stability or minor progression with respect to pleuroparenchymal changes, while 1 showed a marked progression of fibrosis.
Pneumothorax can be recurrent and complicate the course of the disease (Fig. 3). It can be either spontaneous or iatrogenic following interventional procedures or surgery. Treatment is hampered by the presence of bronchopleural fistulae, probably due to the limited healing capability of the affected tissue.6
. This coronal reconstruction shows a right sided pneumothorax and fibrotic infiltrates in both apices (head of arrow).")
Differential radiological diagnosis is performed with the consideration of entities predominantly affecting the upper lobes, such as late-stage sarcoidosis with fibrosis, radiation-induced fibrosis, and apical caps. Apical caps affect geriatric patients with a history of smoking, and functional or radiological worsening is generally absent.26 As the name suggests, they occur in lung apices, while PPFE, although predominantly affecting upper lobes, has a more diffuse subpleural distribution.8
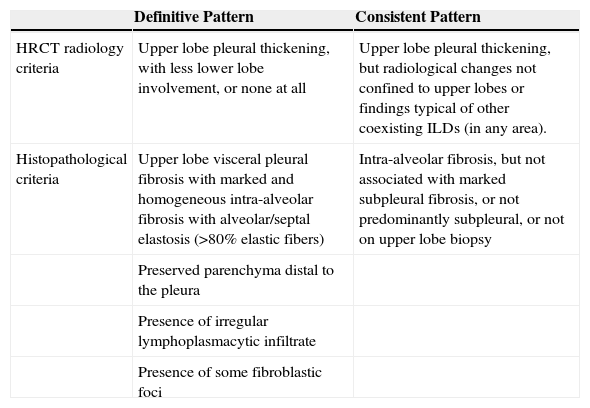
Histological CharacteristicsBased on the similarities between histological and radiological findings in patients with PPFE, some authors have drawn up criteria for establishing either a “definitive” or “consistent with PPFE” pattern11,24,26 (Table 2).
Diagnostic Criteria for Pleuroparenchymal Fibroelastosis (PPFE).
| Definitive Pattern | Consistent Pattern | |
|---|---|---|
| HRCT radiology criteria | Upper lobe pleural thickening, with less lower lobe involvement, or none at all | Upper lobe pleural thickening, but radiological changes not confined to upper lobes or findings typical of other coexisting ILDs (in any area). |
| Histopathological criteria | Upper lobe visceral pleural fibrosis with marked and homogeneous intra-alveolar fibrosis with alveolar/septal elastosis (>80% elastic fibers) | Intra-alveolar fibrosis, but not associated with marked subpleural fibrosis, or not predominantly subpleural, or not on upper lobe biopsy |
| Preserved parenchyma distal to the pleura | ||
| Presence of irregular lymphoplasmacytic infiltrate | ||
| Presence of some fibroblastic foci |
The most relevant histological findings in PPFE are visceral pleural fibrosis and subpleural and parenchymal fibroelastosis. This phenomenon occurs with collagen deposition, extending to the adjacent alveolar walls, mainly in the upper lobes (Fig. 4). In some cases, however, pleural thickening does not occur.11,17,28,29 The transition from normal to injured parenchyma is usually abrupt. There may be few fibroblastic foci, and varying amounts of lymphoplasmacytic infiltrate. Elastosis is not exclusive to PPFE; it can also be found in other IIPs. However, a study comparing lung elastic fibers in a group of IPF patients and a group of PPFE cases found that the latter group presented significantly more fibers.19 Another histological characteristic described in the literature is the co-existence of PPFE with other ILD patterns (UIP, non-specific interstitial pneumonia, hypersensitivity pneumonitis, and even unclassifiable interstitial pneumonia patterns), as discussed above.10,11,26,29 Recently, Oda et al.17 showed poorer survival rates in patients with PPFE and concomitant UIP.
 The alveoli are completely obliterated due to accumulation of elastotic material in the lung wall (hematoxylin and eosin stain). (b) Enlargement of the previous image showing scant lymphoplasmacytic infiltrate. (c) Van Gieson elastic staining highlighting the elastotic material of the wall to show how the alveolar lumen has been replaced by loose connective tissue. (d) Image (b) of the same field under autofluorescence, showing numerous elastic fibers.")
Surgical biopsy of the right upper lobe of a 40-year old female patient with a diagnosis of pleuroparenchymal fibroelastosis. (a) The alveoli are completely obliterated due to accumulation of elastotic material in the lung wall (hematoxylin and eosin stain). (b) Enlargement of the previous image showing scant lymphoplasmacytic infiltrate. (c) Van Gieson elastic staining highlighting the elastotic material of the wall to show how the alveolar lumen has been replaced by loose connective tissue. (d) Image (b) of the same field under autofluorescence, showing numerous elastic fibers.
As discussed above, the presence of concurrent BO and diffuse alveolar damage reported in some cases of pulmonary transplant recipients suggests the PPFE could be a late complication of graft-versus-host-disease.10,24,31–35
No characteristic cell patterns have been described in bronchoalveolar lavage specimens.
Prognosis and TreatmentThe prognostic findings in cases published in the literature vary greatly: some describe a slow, progressive evolution, and other a rapid clinical deterioration.2,11,25,29 As with other IIPs, evolution will depend in part on when diagnosis is made. This is why it is so important to raise awareness of the characteristics of this disease to increase the level of diagnostic suspicion, above all in risk groups (lung and bone marrow transplant recipients or patients with prior chemotherapy treatment). The multidisciplinary team should consider the propensity of these patients for developing pneumothorax. Some authors suggest that an understanding of the clinical and radiological findings associated with PPFE would prevent the need for surgical biopsy.28 It would be interesting to evaluate the yield of other diagnostic techniques, such as cryobiopsy, since these have the potential to minimize risks and provide a good histopathological diagnosis.
At present, no effective treatment has been found for PPFE. Patients have been treated empirically with corticosteroids, N-acetyl cysteine and several different immunosuppressants, although none have given clear evidence of improvement.11,29 Oxygen therapy is indicated in patients with respiratory failure, and lung transplantation is an option in patients meeting transplant criteria. Our group recently reported the case of a successful transplant in a PPFE patient, with 24 months survival to date (unpublished data).
Further multicenter studies, international registries and studies in animal models are needed to improve understanding of the natural history of PPFE, its pathogenesis and trigger factors. This may show that PPFE is more common than initially thought, and enable clinicians to establish early diagnosis strategies and appropriate treatment regimens.
FundingKarina Portillo Carroz has received a research grant from the Sociedad Española de Neumología y Cirugía Torácica (2013) and also an EPID – futuro grant from Roche (2013).
Conflict of InterestsThe authors declare they have no conflicts of interest.
We would like to thank the members of the Grup d’Estudi Clínic Radiològic i Anatomopatològic de les Malalties Pulmonars Intersticials Difuses (CRAMPID) team for their ongoing encouragement, and also Dr. María Teresa Fernández Figueras for her invaluable help and permission to publish her histopathological images.
Please cite this article as: Portillo K, Guasch Arriaga I, Ruiz-Manzano J. Fibroelastosis pleuropulmonar: ¿es también una entidad idiopática? Arch Bronconeumol. 2015;51:509–514.







