
We present three cases of two unrelated families with cystic fibrosis (CF) confirmed diagnosis, high chloride concentrations, mild clinical phenotype and the synonymous CFTR A209= variant linked to a deep intronic variant affecting splicing, and in compound heterozygosity with another CFTR-causing variant. These findings underscore the need for deeper investigation of the synonymous variant, as its benign appearance might obscure its pathogenic role linked to the intronic variant.
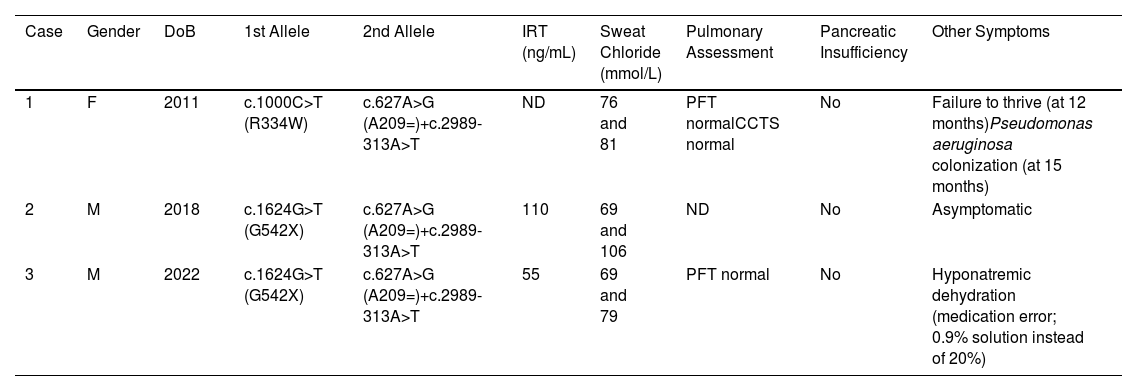
Two maternal cousins (cases 1 and 2) and an unrelated male infant (case 3) were diagnosed with CF (Table 1). Case 1 initially presented with failure to thrive, later developing transient Pseudomonas aeruginosa colonization but has remained asymptomatic since. Case 2, diagnosed through neonatal screening, showed a mild phenotype without complications. Case 3, also diagnosed via neonatal screening, experienced a single episode of hyponatremic dehydration due to a medication error, with no further symptoms observed. All three cases demonstrate a mild clinical course, contrasting with the severity often associated with CF.
Demographic, Genotyping and Clinical Data of the Three Reported Patients.
| Case | Gender | DoB | 1st Allele | 2nd Allele | IRT (ng/mL) | Sweat Chloride (mmol/L) | Pulmonary Assessment | Pancreatic Insufficiency | Other Symptoms |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 2011 | c.1000C>T (R334W) | c.627A>G (A209=)+c.2989-313A>T | ND | 76 and 81 | PFT normalCCTS normal | No | Failure to thrive (at 12 months)Pseudomonas aeruginosa colonization (at 15 months) |
| 2 | M | 2018 | c.1624G>T (G542X) | c.627A>G (A209=)+c.2989-313A>T | 110 | 69 and 106 | ND | No | Asymptomatic |
| 3 | M | 2022 | c.1624G>T (G542X) | c.627A>G (A209=)+c.2989-313A>T | 55 | 69 and 79 | PFT normal | No | Hyponatremic dehydration (medication error; 0.9% solution instead of 20%) |
DoB: date of birth; TIR: immunoreactive trypsinogen; ND: not determined; NA: not applicable; PFT: pulmonary function test; CCTS: chest CT scan.
Genetic analysis of the CFTR gene was performed using genomic DNA extracted from blood samples. Targeted enrichment of exonic and flanking intronic regions was followed by massive parallel sequencing (NextSeq platform) to detect coding and intronic variants. To confirm the presence of the c.2989-313A>T variant, PCR amplification of exon 18 was performed, followed by Sanger sequencing (AB3500 Genetic Analyzer, Applied Biosystems). Variant analysis was conducted in both index cases and their parents to assess segregation patterns and confirm the inheritance of the identified variants. The synonymous NM_000492.4:c.627A>G (p.Ala209=) variant was identified in linkage disequilibrium with the NM_000492.4:c.2989-313A>T and in compound heterozygosity with another pathogenic variant. mRNA analysis showed that the c.627A>G variant did not affect transcription.
The c.627A>G variant was first reported in 2001 by Le Maréchal et al.1 as having no impact on the amino acid sequence in a CF patient. In 2019, Bergougnoux et al.2 linked it to the deep intronic c.2989-313A>T variant, identifying it as a complex allele with a broad phenotypic spectrum ranging from mild chronic bronchitis to severe CF symptoms. In contrast, the three cases presented consistently exhibit a mild phenotype, with normal pulmonary function, no pancreatic insufficiency, and no significant health impacts, suggesting that the intronic variant may lead to milder clinical manifestations in these patients. Given their favorable clinical course, none of them have required CFTR modulators.
While Bergougnoux et al. provided data on symptom onset, they did not specify the exact age of diagnosis, making it difficult to assess whether these patients had access to early detection or long-term follow-up. In contrast, our patients were diagnosed within the first year of life due to newborn screening or early clinical suspicion, allowing for timely intervention and continuous monitoring, which may have influenced their milder presentation. Additionally, the treatment regimens received by the 2019 cohort were not explicitly documented, making it difficult to determine the impact of clinical management and healthcare accessibility on disease progression. This raises the question of whether the milder phenotype observed in our cohort is primarily due to early diagnosis and proactive medical care or whether other genetic or environmental modifiers contribute to the observed differences.
The c.627A>G variant, while considered benign, gains significance due to its consistent linkage disequilibrium with the pathogenic intronic c.2989-313A>T variant. This deep intronic variant generates a pseudo-exon, leading to a premature stop codon and potentially truncated CFTR protein,2 as seen in all three cases. Its co-occurrence with the synonymous variant highlights the need for thorough genetic investigation, as routine gene analysis often excludes intronic regions.
Although mRNA analysis is a direct method for characterizing splicing variants,3 its limitations in accessibility and sample stability make it less practical.4 For detecting the c.2989-313A>T variant linked to c.627A>G, cost-effective methods like Sanger sequencing are sufficient and more feasible for routine diagnostics.
Statement of EthicsThe authors have no ethical conflicts to disclose.
Contribution of Each AuthorFMB: Conception and study design, data acquisition, data analysis, manuscript drafting, and final manuscript approval.
GGR: Data analysis, manuscript drafting, and final manuscript approval.
CPM: Writing – review and editing, and final manuscript approval.
OMM: Writing – review and editing, and final manuscript approval.
ACC: Writing – review and editing, and final manuscript approval.
All authors have contributed substantially to obtaining the results and preparing the manuscript in accordance with the ICMJE criteria.
Artificial Intelligence InvolvementNo part of this manuscript has been produced with the help of artificial intelligence software or tools.
Funding of the ResearchThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors have not received any funding specifically for this correspondence.
Conflicts of InterestThe authors declare not to have any conflicts of interest that may be considered to influence directly or indirectly the content of the manuscript.
The authors have no conflicts of interest to declare.







