


Idiopathic pulmonary fibrosis (IPF) is a chronic and progressive fatal fibrotic interstitial lung disease of unknown etiology characterized by progressive deposition of extracellular matrix and destruction of alveolar architecture.1 It results in respiratory failure and patients have a poor prognosis, the median survival being about 3–5 years following diagnosis.2 Lung transplantation is the only curative treatment for IPF. However, notable advances have been made in its pharmacological therapy during the last 5 years with the development of new agents designed to inhibit the processes that drive fibrosis.3 Promising results have recently been made public from phase I and II clinical trials with the novel targets of the autotaxin-lysophosphatidic acid (ATX/LPA) pathway,4,5 and the transforming growth factor-β (TGF-β) pathway6,7 that are hypothesized to be central in the development of IPF.8,9 Thus, the ATX inhibitor GLPG1690 was generally well tolerated and exhibited a safety profile similar to placebo, demonstrating favorable effects on mean change from baseline in forced vital capacity (FVC) at week 12 compared to placebo (25mL vs −70mL).5 Two phase III trials are currently ongoing.10 In the case of TGF-β suppression, GSK3008348 an αvβ6 integrin inhibitor administered as an inhaled solution, was also well tolerated with no reports of serious adverse events or clinically significant abnormalities that were attributable to study treatment.6 Due to the increasing interest triggered by the new agents, reliable serum biomarkers would help to improve both diagnostic approach and monitoring of drug effects. Thus, serum levels of surfactant protein A (SP-A), a pulmonary collectin, have been shown to be useful in predicting prognosis or monitoring disease activity.11,12 Even changes in serum SP-A levels could reflect the outcomes of anti-fibrotic drug therapy with pirfenidone or nintedanib.13 The usefulness of serum biomarkers in patients undergoing anti-fibrotic drug therapy based on (ATX/LPA) and TGF-β pathways is not well characterized. The purpose of this study is to assess whether the pro-fibrotic compounds ATX, LPA and TGF-β, together with SP-A, could serve as potential biomarkers to monitor the therapeutic response of these novel IPF treatments.
We here report the results of a single-center study consisted of the measurement of serum levels of pro-fibrotic markers in 29 IPF confirmed patients recruited from the Interstitial Lung Diseases Unit at the Pneumology Service at University Hospital of Malaga ‘Virgen de la Victoria’ during 56 months. Thirty healthy individuals were included as comparison group and paired with sex, age, and smoking status with IPF patients. The local ethics committee approved this study. Prior to enrolment, participants were informed about the study, and provided written informed consent. A multidisciplinary group of Pneumologists, Radiologists and Pathologists established and discussed the diagnostics of IPF cases following the European Respiratory Society/American Thoracic Society/Japanese Respiratory Society/Latin American Thoracic Society criteria.1
The serum levels markers in IPF patients were compared with the levels found in healthy volunteers. The study included a single visit of participants. After providing the informed consent, a pulmonary function test was performed and a whole blood sample was drawn from a vein (venipuncture), and collected after fasted for 8h in tubes containing EDTA (50mM). For plasma preparation, blood was centrifuged at 1200×g for 10min at 4°C and the supernatant was stored in siliconized tubes at −80°C until use. The IPF treatments at the time of venipunture were recorded. Thus, 11 (37.9%) patients were treated with N-acetylcysteine as monotherapy, 3 (10.3%) with systemic corticosteroids and 1 (3.4%) with monoclonal antibodies. In 8 (27.6%), these drugs were taken in combination, together with immunosuppressive drugs (azathioprine or cyclophosphamide) in 3 (10.3%) patients. Levels of ATX, LPA, SP-A and TGF-β1 in serum were measured by sandwich-type enzyme-linked immunosorbent assays, using commercially available ELISA kits (Autotaxin K-5600, LPA K-2800S, Echelon®; SP-A NBP2-76692, Novus Biologicals; TGF-β1 SEA124Hu, Cloud-Clone Corp) according to the manufacturer's instructions. To compare quantitative measures (mean±SD), t-test or Mann–Whitney U test were used, while chi-square test was used for qualitative (%). The receiver-operating characteristic (ROC) curves and Youden index were applied to determine the optimal cut-off of serum markers to distinguish IPF from healthy participants. A significance of 5% (P<.05) was required to consider a difference to be statistically significant. Statistical analysis was performed using SPSS software (version 24.0; IBM SPSS Statistics, Chicago, IL).
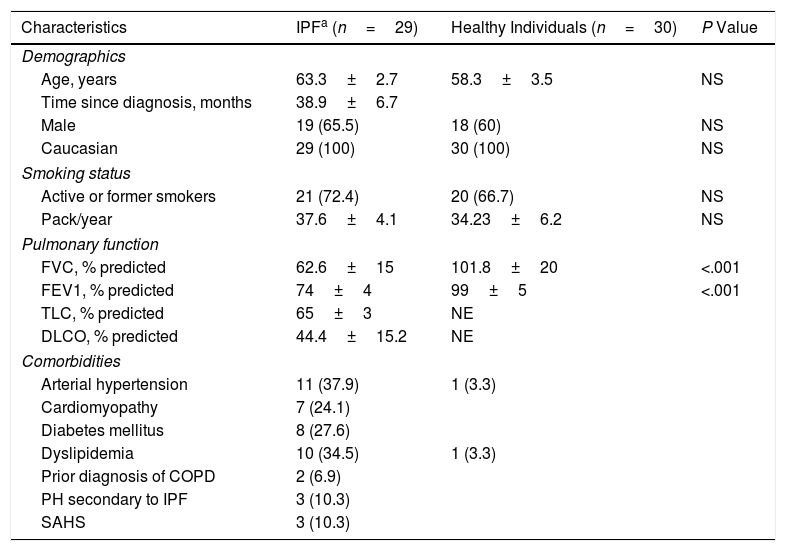
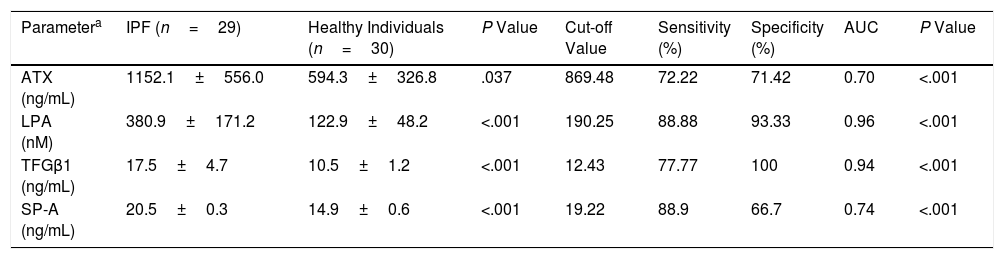
These two populations showed similar demographics and smoking status (Table 1) whereas the functional parameters (% predicted) FVC and FEV1 (forced expiratory volume in 1 second) in IPF patients were statistically significantly lower than those in comparison group (Table 1, P<.001). However, the serum levels of the pro-fibrotic markers in IPF patients were all statistically significantly higher than those in healthy participants. The IPF patients showed a clear increase of 66.66% of TGF-β1 plasma levels (+7.05ng/mL), two-fold more abundant the values of ATX (+557.77ng/mL), and three-fold the LPA values (+257.91nM) (Table 2). Similarly, a cohort study found two-fold more abundant lysophosphatidylcholine (a LPA precursor) in IPF patients compared to healthy participants, being proposed as biomarker.14 According to the ROC curve analyses, the sensitivities and specificities estimated of each biomarker are shown in Table 2. The serum biomarkers values in IPF patients were all higher than the optimal cut-off values of ATX, LPA and TGF-β1 (869.48ng/mL, 190.25nM and 12.43ng/mL, respectively); whereas the serum biomarkers values in healthy participants were lower (Table 2). The areas under the ROC curves (AUC) for IPF patients in comparison with control participants were all ≥0.70 (P<.001), showing LPA and TGF-β1 better AUC profiles to distinguish IPF from healthy individuals (>0.90). Our study also demonstrated a statistically significant increase of SP-A serum levels in IPF patients of 37.58% compared to control (P<.001), being higher than the optimal cut-off values found (Table 2). The SP-A AUC (0.74) was similar to ATX AUC, lower than LPA and TGF-β1 AUC profile, which showed the largest AUC for distinguishing IPF from healthy participants (0.96 and 0.94, respectively). Limitations: first, our work is a single-center study, which limits its external validity. Second, the small sample size does not allow studying the effect of different variables (tobacco with a high percentage of smokers, age or time since diagnosis) on the levels of these biomarkers. In conclusion, changes in these serum biomarkers may serve as useful tools for monitoring the novel IPF anti-fibrotic drugs, providing novel insights for further investigations to verify the clinical relevance of these serum biomarkers, mainly LPA and TGF-β, in the context of anti-fibrotic therapies for IPF.
Baseline Characteristics of the Patients in Terms of Demographics, Smoking Status, Pulmonary Function and Comorbidities.
| Characteristics | IPFa (n=29) | Healthy Individuals (n=30) | P Value |
|---|---|---|---|
| Demographics | |||
| Age, years | 63.3±2.7 | 58.3±3.5 | NS |
| Time since diagnosis, months | 38.9±6.7 | ||
| Male | 19 (65.5) | 18 (60) | NS |
| Caucasian | 29 (100) | 30 (100) | NS |
| Smoking status | |||
| Active or former smokers | 21 (72.4) | 20 (66.7) | NS |
| Pack/year | 37.6±4.1 | 34.23±6.2 | NS |
| Pulmonary function | |||
| FVC, % predicted | 62.6±15 | 101.8±20 | <.001 |
| FEV1, % predicted | 74±4 | 99±5 | <.001 |
| TLC, % predicted | 65±3 | NE | |
| DLCO, % predicted | 44.4±15.2 | NE | |
| Comorbidities | |||
| Arterial hypertension | 11 (37.9) | 1 (3.3) | |
| Cardiomyopathy | 7 (24.1) | ||
| Diabetes mellitus | 8 (27.6) | ||
| Dyslipidemia | 10 (34.5) | 1 (3.3) | |
| Prior diagnosis of COPD | 2 (6.9) | ||
| PH secondary to IPF | 3 (10.3) | ||
| SAHS | 3 (10.3) | ||
Continuous variables are expressed as the mean (± SD). Categorical data are expressed as n (%). Abbreviations: IPF, idiopathic pulmonary fibrosis; FVC, forced vital capacity; FEV1, forced expiratory volume in 1 second; TLC, total lung capacity; DLCO, diffusing capacity for carbon monoxide; COPD, chronic obstructive pulmonary disease; PH, pulmonary hypertension; SAHS, sleep apnea-hypopnea syndrome; NS, non-significant; NE, not evaluated.
Serum Levels Markers and ROC Curves Analysis to Distinguish IPF From Healthy Participants.
| Parametera | IPF (n=29) | Healthy Individuals (n=30) | P Value | Cut-off Value | Sensitivity (%) | Specificity (%) | AUC | P Value |
|---|---|---|---|---|---|---|---|---|
| ATX (ng/mL) | 1152.1±556.0 | 594.3±326.8 | .037 | 869.48 | 72.22 | 71.42 | 0.70 | <.001 |
| LPA (nM) | 380.9±171.2 | 122.9±48.2 | <.001 | 190.25 | 88.88 | 93.33 | 0.96 | <.001 |
| TFGβ1 (ng/mL) | 17.5±4.7 | 10.5±1.2 | <.001 | 12.43 | 77.77 | 100 | 0.94 | <.001 |
| SP-A (ng/mL) | 20.5±0.3 | 14.9±0.6 | <.001 | 19.22 | 88.9 | 66.7 | 0.74 | <.001 |
ATX: autotaxin; LPA: lysophosphatidic acid; TGF-β1: transforming growth factor-β1; SP-A: surfactant protein A; AUC: area under the curve.
The values were detectable in all samples analyzed. ELISA kits, sensitivity limits: Autotaxin K-5600 (3.21ng/mL, Intra-Assay: CV 3%, Inter-Assay: CV 9%); LPA K-2800S (24nM, Intra-Assay: 6.5%–8.7%, Inter-Assay: 8.9%–15.33%); SP-A NBP2-76692 (9.375pg/mL, Intra-Assay: 5.03%, Inter-Assay: 4.83%); TGF-β1 SEA124Hu (6.3pg/mL, Intra-Assay: CV<10%, Inter-Assay: CV<12%).
The purchase of necessary laboratory reagents was supported in part by Almirall, S.A. The company had no role in the study design, the collection, analysis, or interpretation of data, the writing of the report, or the decision to submit the article for publication.







