



Introducción
La enfermedad pulmonar obstructiva crónica (EPOC) es un problema sanitario de primer orden debido a su importante morbimortalidad en quienes la padecen y el enorme gasto sanitario que implica1. Según la GOLD2 (Global Initiative for the Diagnosis, Management and Prevention of Obstructive Lung Diseases), se define como una enfermedad caracterizada por obstrucción crónica al flujo aéreo, que habitualmente es progresiva y se asocia a una respuesta inflamatoria anormal a la inhalación de partículas o gases tóxicos. Existen pocas dudas actualmente de que el humo del tabaco es el principal factor desencadenante de esta reacción inflamatoria en el pulmón de los fumadores. Sin embargo, los mecanismos por los cuales el tabaco desencadena la enfermedad y los responsables de que la respuesta inflamatoria continúe al dejar de fumar están todavía lejos de ser comprendidos por completo, y por lo tanto el manejo de la enfermedad es esencialmente sintomático.
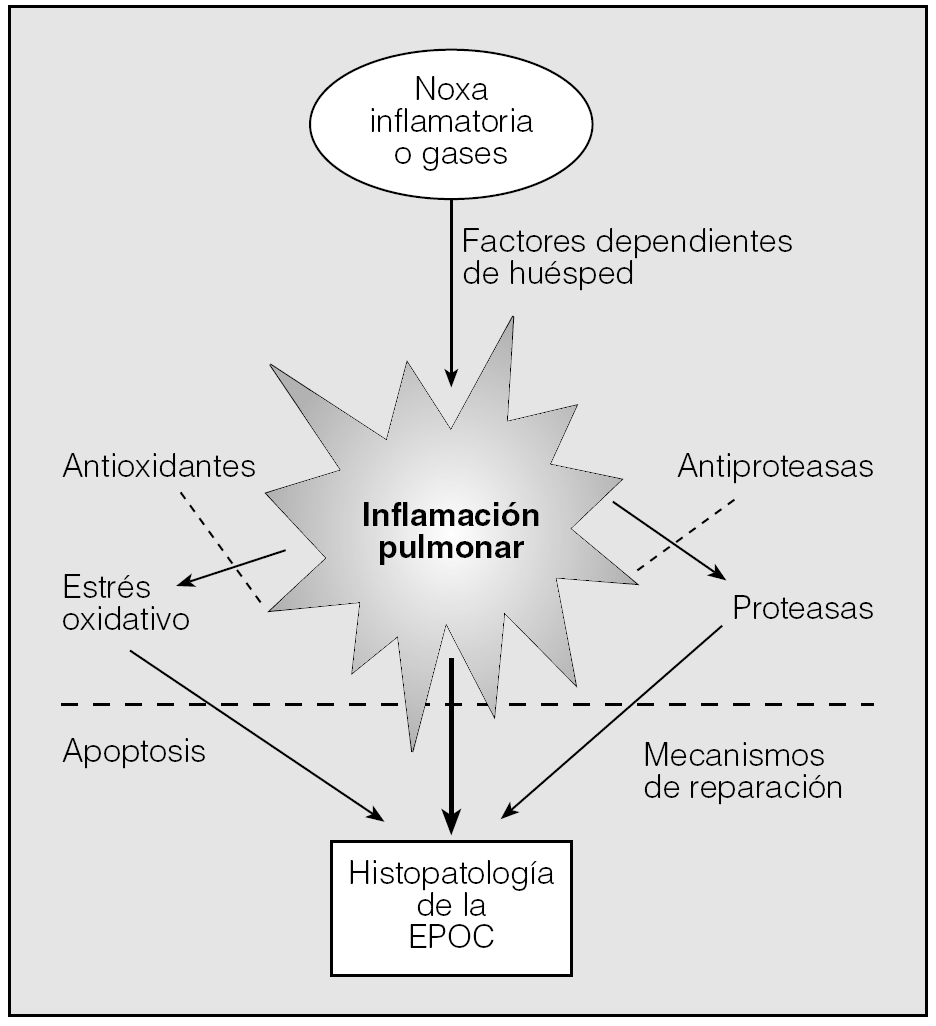
El conocimiento de la etiopatogenia de la EPOC ha avanzado considerablemente en los últimos años. Desde los tiempos de Laennec hasta casi la primera mitad del siglo xx predominaban las explicaciones mecánicas del enfisema, sin considerarse el efecto del tabaco. A finales de los años 1950 Liebow propone un modelo de atrofia vascular en el enfisema (recientemente reproducido por Voelkl et al en un modelo animal3) y más tarde surge la hipótesis del desequilibrio entre proteasas y antiproteasas. Esta hipótesis de la patogenia del enfisema se basa en dos observaciones muy importantes: por una parte, Laurell y Eriksson describen la asociación entre el enfisema y el déficit de alfa-1-antitripsina; por otra, el grupo de Gross describe el primer modelo de enfisema inyectando una proteasa vegetal, la papaína, en los pulmones de animales de experimentación. La integración de esta hipótesis a las observaciones de la naturaleza inflamatoria de la enfermedad puede sintetizarse en los siguientes puntos: a) la exposición crónica al humo del tabaco recluta células inflamatorias a los espacios aéreos del pulmón; b) estas células inflamatorias liberan mediadores inflamatorios con capacidad elastolítica, que degradan la matriz extracelular, y c) los mecanismos de reparación alveolar están alterados, lo que da lugar a la destrucción de los espacios alveolares característicos del enfisema pulmonar. La integración de estos hallazgos, junto con el papel del estrés oxidativo y el desequilibrio entre proteasas y antiproteasas, es la base que sustenta el conocimiento actual de la etiopatogenia de la EPOC (fig. 1).

Fig. 1. Etiopatogenia de la EPOC. (Modificada de Pauwels et al2.)
La reacción inflamatoria al humo del tabaco aparece en todos los fumadores, pero sólo un 15-20% desarrollará la enfermedad. Hay factores predisponentes, tanto ambientales como genéticos, que están aún por determinarse.
A pesar de la evidencia de estos hechos y de la enorme importancia tanto de la incidencia de la EPOC como del consumo de recursos que genera, la investigación sobre los mecanismos celulares y moleculares, así como sobre nuevos tratamientos para la enfermedad, no se ha desarrollado de forma importante hasta las últimas dos décadas, probablemente debido a que no se comprendía la naturaleza inflamatoria de la enfermedad y, cuando se comprendía, se interpretaba como similar a la observada en el asma. Del mismo modo, el hecho de que en una mayoría de estos pacientes el desarrollo de la enfermedad sea consecuencia del consumo prolongado de tabaco, hacía que se considerara que era "culpa" del paciente. Finalmente, la ausencia de un modelo animal que reproduzca los cambios característicos de la EPOC tras la exposición al humo del tabaco ha sido determinante en este retraso. De hecho, no ha sido hasta la reciente iniciativa GOLD cuando en la definición de EPOC se ha reconocido la importancia de la asociación de la obstrucción crónica e irreversible al flujo aéreo característica de estos pacientes con el concepto de inflamación persistente en respuesta a una noxa inhalada2.
Este trabajo propone revisar los avances recientes en el conocimiento de los diferentes aspectos etiopatogénicos de la enfermedad y los enfoques terapéuticos que pueden plantearse para abordar la EPOC bajo el prisma de los descubrimientos del laboratorio.
Etiopatogenia de la EPOC
Factores genéticos
La observación de que sólo aproximadamente un 20% de los fumadores desarrollará una pérdida acelerada de función pulmonar invita a pensar que existen factores genéticos que influyen en el desarrollo de la enfermedad. La evidencia de agregación familiar en pacientes con inicio temprano de la EPOC4 y la diferencia de prevalencia entre distintos grupos raciales sustentan la idea de una predisposición genética. En pacientes con déficit de alfa-1-antitripsina y fenotipo PiZZ, con títulos de la enzima inferiores al 10% de los valores normales, se produce un desarrollo temprano de enfisema pulmonar, que se ve acelerado por el hecho de fumar, lo que indica una clara predisposición genética para el desarrollo de la EPOC. La alfa-1-antitripsina es una serpina con acción muy selectiva contra la elastasa de los neutrófilos. Sin embargo, sólo un 1% de los pacientes con EPOC tienen déficit de alfa-1-antitripsina, lo que ha llevado a buscar asociaciones entre EPOC y polimorfismos de otros genes que pudieran intervenir en su fisiopatología. Otras antiproteasas, como las metaloproteasas de la matriz (MMP), han sido implicadas en modelos animales de EPOC5, pero los polimorfismos en estos genes todavía no han mostrado un papel importante en humanos. No obstante, otras serpinas pueden tener un efecto en la EPOC, especialmente las que se expresan en el epitelio de la vía aérea, como la serpina-2, que es un inhibidor del plasminógeno tisular que se ha implicado por cartografía genética en el cromosoma 2q33-35. Se han observado asociaciones entre el polimorfismo en el promotor de la región del gen que codifica el factor de necrosis tumoral alfa (TNF-α), y que produce un aumento de la producción de esta citocina, y un riesgo 10 veces mayor de desarrollar EPOC en una población tailandesa. Otros estudios han encontrado una asociación entre una variante polimórfica de la hidrolasa epóxido microsómica enzima que interviene en el metabolismo de los epóxidos generados por el humo del tabaco y un riesgo 5 veces mayor de desarrollar EPOC. También se ha implicado a los antioxidantes, como la hemeoxigenasa-1, aunque los resultados no han sido reproducidos. El factor transformador del crecimiento β1, una importante quimiocina antiinflamatoria y profibrótica, también se ha asociado con la EPOC en diversos trabajos6. Los resultados de numerosos estudios que indican la relevancia potencial de la apoptosis3 en el desarrollo de enfisema hacen posible que los polimorfismos en genes reguladores de este proceso puedan ser relevantes. Asimismo, la desregulación de las histonas deacetilasas en la EPOC hace que estos genes sean posibles candidatos de estudio7.
El reciente desarrollo de nuevas técnicas de genética molecular, como los chips génicos, permitirá en el futuro próximo conocer qué genes están sobreexpresados o son polimórficos, lo que será de utilidad no sólo para identificar a pacientes con riesgo, sino también para descubrir moléculas diana donde investigar futuros tratamientos8.
Tratamiento génico
La sustitución de genes defectuosos o la administración transitoria de genes que afectan a la función o modulan respuestas celulares son abordajes novedosos para tratar enfermedades respiratorias que están ganando credibilidad. Los vectores más comúnmente utilizados para la transfección de genes son los lentivirus, adenovirus de replicación deficiente y los liposomas/ complejos ADN sintéticos. En los casos de un único defecto genético, como el déficit de alfa-1-antitripsina, todavía no está claro si el reemplazo del gen defectuoso puede alterar el curso de la enfermedad. La mayoría de los intentos realizados han sido infructuosos por el corto tiempo de expresión del gen y las elevadas concentraciones requeridas para la eficacia terapéutica. En la actualidad no existe una investigación en genoterapia para la EPOC, aunque son concebibles varias posibilidades; por ejemplo, el reestablecimiento del desequilibrio entre proteasas y antiproteasas, o bien la sobreexpresión de genes de antiproteasas como el de la alfa-1-antitripsina o el del inhibidor de la secreción de proteasa leucocitaria (SLPI). También podría dirigirse a las moléculas de adhesión de los neutrófilos para reducir el aflujo de células inflamatorias en el parénquima pulmonar.
La genoterapia, aunque todavía en fases muy tempranas de desarrollo, es un enfoque prometedor para el tratamiento de la EPOC, especialmente en los casos de déficit genético único, y precisa mejoras en los sistemas vectoriales de transfección de genes9.
Inflamación en la EPOC
La inflamación pulmonar puede encontrarse en la mayoría de los fumadores, según se ha demostrado en varios estudios que analizan el parénquima pulmonar, al igual que en otros que se centran en biopsias bronquiales o esputo inducido. Sin embargo, parece que la respuesta inflamatoria exagerada a la inhalación de partículas o gases, más allá de la normal respuesta inflamatoria protectora al humo del tabaco, es un hecho característico de la EPOC que acaba produciendo daño en el pulmón de los fumadores susceptibles1. Estudios recientes comienzan a caracterizar el tipo, lugar y grado de inflamación en el pulmón de los pacientes con EPOC, así como su relación con la intensidad de la enfermedad10.
Estudios de biopsias bronquiales de pacientes con EPOC leve o moderada muestran una mayor infiltración por células inflamatorias en las vías aéreas centrales de dichos pacientes, comparados con no fumadores y fumadores que no desarrollan la enfermedad. En la mucosa bronquial de los pacientes con EPOC predominan los linfocitos T, principalmente CD8+, y los macrófagos (CD68+). Se ha señalado que la presencia de linfocitos T podría diferenciar entre los fumadores que desarrollan EPOC y los que no la desarrollan, debido a que se ha hallado una relación entre el número de células T, la cantidad de destrucción alveolar y la importancia de la obstrucción al flujo aéreo11. Estudios en muestras quirúrgicas de parénquima pulmonar encontraron que la progresión de la EPOC estaba estrechamente asociada a un mayor volumen de tejido en las paredes bronquiales y a una acumulación de exudados inflamatorios en la luz de las pequeñas vías aéreas, lo que constituye un argumento de peso en contra de las teorías que consideran la EPOC avanzada como una enfermedad "seca". El perfil celular se correspondía con el previamente descrito, con una mayor cantidad de neutrófilos, macrófagos y linfocitos T CD8+ y CD4+, pero además hay una mayor cantidad de células B y de folículos linfoides peribronquiales conforme progresa la enfermedad10, lo que confirma la participación de la inmunidad innata y adaptativa en la patogenia de la EPOC.
El mecanismo por el cual las células T CD8+ se acumulan en el pulmón de los pacientes con EPOC no se conoce del todo. Las células T en las vías aéreas periféricas de dichos pacientes muestran una expresión aumentada de CXCR3, un receptor activado por la proteína 10 inducible por interferón, y la expresión de dicha proteína 10 está aumentada en células del epitelio bronquial, lo que podría contribuir a la acumulación de células CD8+ que expresan preferentemente CXCR3. También hay un aumento de las células T CD4+ en pacientes con EPOC, sobre todo en fases avanzadas. Es posible que la colonización crónica por patógenos bacterianos o víricos sea responsable de la respuesta inflamatoria amplificada de la EPOC12. Es también posible que el propio humo del tabaco dañe las células epiteliales bronquiales y genere nuevos autoantígenos que desencadenen la respuesta inmunológica e inflamatoria, y se ha llegado a postular la posibilidad de que la EPOC sea una enfermedad autoinmunitaria13,14. Tampoco se conoce con precisión qué papel desempeñan los linfocitos T, aunque se conjetura que podrían activar diversas vías apoptóticas a través de la liberación del TNF-α, perforinas y granzimas.
Los neutrófilos también se encuentran aumentados en el esputo de los pacientes con EPOC15. Los neutrófilos tienen la capacidad de secretar proteinasas, incluidas la elastasa de los neutrófilos, catepsina G, proteinasa 3, así como MMP-8 y MMP-9. Estas proteinasas pueden contribuir a la destrucción alveolar y son potentes estimulantes de la secreción de moco. El número de neutrófilos encontrados en biopsias bronquiales y esputo inducido se ha relacionado con la gravedad de la EPOC16, así como con la rapidez de la pérdida de función pulmonar. El humo del tabaco puede ser responsable del aumento de la cantidad de neutrófilos circulantes y de la modificación de su deformabilidad para secuestrarlos en los capilares del pulmón17, así como tener un efecto directo sobre la producción de granulocitos en la médula ósea a través de la liberación del factor estimulante de colonias granulocíticas y microcíticas producido por los macrófagos. Una vez secuestrados, los neutrófilos se adhieren a las células endoteliales y migran al tracto respiratorio bajo el control de factores quimiotácticos como el leucotrieno B4 o la interleucina (IL) 8.
El número de macrófagos es hasta 10 veces mayor en las vías aéreas, parénquima y lavado broncoalveolar de pacientes con EPOC. Además, también se relaciona con la gravedad de la EPOC12. El humo del tabaco activa los macrófagos, y éstos liberan mediadores inflamatorios como el TNF-α, la IL-8, la proteína quimiotáctica de los monocitos-1 (MCP-1), el leucotrieno B4 o especies reactivas de oxígeno, así como proteasas tales como MMP-2, MMP-9, MMP-12, y catepsinas K, L y S. Los pulmones de los fumadores sin EPOC también muestran mayor número de macrófagos, probablemente secundario a la liberación de quimiocinas quimiotácticas de los monocitos como la MCP-1. Sin embargo, los macrófagos en los pacientes con EPOC están más activados, liberan más proteínas inflamatorias y tienen mayor capacidad elastolítica que los de fumadores sin EPOC18.
Igualmente hay un número mayor de células dendríticas en el pulmón de pacientes con EPOC, sobre todo en las paredes alveolares y vías aéreas. Todavía no se conoce su función, aunque podrían tener un papel relevante en la respuesta inmunitaria innata y adaptativa19.
Las células epiteliales también tienen un papel muy importante en este proceso inflamatorio, ya que son activadas directamente por el humo del tabaco y liberan mediadores (factor estimulante de colonias granulocíticas y microcíticas, IL-8, IL-1β, TNF-α) que inician la cascada inflamatoria. También pueden ser fuente de antioxidantes y transportar inmunoglobulina-alfa, desempeñando por tanto un papel en la inmunidad adaptativa.
Se han realizado importantes avances en el conocimiento de los mecanismos moleculares del proceso inflamatorio de la EPOC. Muchos de los mediadores inflamatorios expresados en la EPOC están controlados por factores de transcripción como el factor nuclear-κB (NF-κB). Este factor es necesario para la transcripción de muchas proteínas inflamatorias y forma parte de un complejo activador de la cromatina celular, por el que se consigue cambiar la configuración de ésta y hacerla activa mediante un proceso de acetilación. Un mecanismo regulador de este proceso es la desacetilación, que al volver a conformar la cromatina en su posición de reposo o inactiva frena la transcripción inflamatoria. Recientemente se ha demostrado que el humo del tabaco es capaz de alterar este mecanismo regulador de la transcripción celular20 y que los pacientes con EPOC tienen una capacidad menor de desacetilación medida como actividad de la enzima histona desacetilasa (HDAC) y, por tanto, un estado de la cromatina más activo que permite una mayor transcripción de genes inflamatorios. Esta disminución de la capacidad desacetiladora (actividad HDAC) se relaciona significativamente con la gravedad de la enfermedad medida por el volumen espiratorio forzado en el primer segundo7.
Inflamación durante las exacerbaciones de la EPOC
Aunque se da por sentado que las exacerbaciones de la EPOC se asocian con una mayor inflamación bronquial, a menudo es difícil de demostrar por la dificultad en la toma de muestras histológicas en estos pacientes. Los fumadores y los pacientes con EPOC estable tienen una respuesta inflamatoria en todo el árbol traqueobronquial que se caracteriza por la infiltración de macrófagos y linfocitos CD821. Sin embargo, este patrón inflamatorio cambia durante las exacerbaciones, con un incremento del número de neutrófilos en la vía aérea, y en los pacientes con EPOC leve puede encontrarse aumento del número de eosinófilos22. Estudios en esputo inducido23 indican que hay un aumento de la producción de citocinas inflamatorias, particularmente IL-6 e IL-8, que es un potente quimioatrayente de neutrófilos. El mecanismo celular de esta inflamación neutrofílica es poco conocido. Un estudio reciente demuestra que la activación y atracción de neutrófilos al pulmón de pacientes con EPOC grave están reguladas por la activación de las vías CXCL5 y CXCL8, posiblemente inducidas por agentes víricos. El papel de la colonización y del cambio de cepas de Haemophilus influenzae durante la agudización y su relación con la inflamación están siendo también motivo de estudio24.
Tratamiento antiinflamatorio
Glucocorticoides
Los glucocorticoides son poco efectivos a la hora de controlar la inflamación crónica que subyace en la etiopatogenia de la EPOC. Varios estudios han demostrado que no suprimen ni las células, ni las citocinas ni las proteasas implicadas en su desarrollo25,26. Los mecanismos están todavía por aclarar, aunque se han producido importantes avances en los últimos años. Los glucocorticoides prolongan la supervivencia de los neutrófilos, lo que contribuye a la inflamación neutrofílica característica de la EPOC. Aunque algunos autores han postulado que el estrés oxidativo puede disminuir la translocación nuclear del receptor glucocorticoide (RG)27, este hecho no se ha confirmado en estudios posteriores28. Otro efecto demostrado del humo del tabaco es la disminución de la actividad de las HDAC, lo que podría explicar, al menos en parte, la resistencia a los efectos antiinflamatorios de los glucocorticoides en pacientes con EPOC y asmáticos fumadores20. Las estrategias terapéuticas encaminadas a desbloquear este mecanismo de resistencia podrían llegar a hacerse posibles, por ejemplo, en forma de fármacos que aumenten la acti-vidad de la HDAC a fin de volver a sensibilizar las células a los efectos de los glucocorticoides. La teofilina a dosis bajas ha demostrado ser capaz de potenciar la actividad de la HDAC in vitro y ex vivo en pacientes con EPOC, y aumentar la capacidad antiinflamatoria de éstos29. La asociación de glucocorticoides inhalados a agonistas beta de larga acción parece ser capaz depotenciar los efectos antiinflamatorios de los prime-ros a través de una mayor translocación nuclear del RG, y ha demostrado una mayor disminución de los neutrófilos en esputo y células T en biopsias bronquiales30.
Glucocorticoides disociados
Muchos de los efectos antiinflamatorios de los glucocorticoides se deben a la inhibición de los factores de transcripción (transrepresión), mientras que los efectos endocrinológicos y metabólicos están mediados por la unión a los elementos de respuesta glucocorticoide del ADN (transactivación). Este hecho ha llevado a los investigadores a buscar moléculas de glucocorticoides que tengan una función selectiva transrepresora, para evitar así los efectos secundarios debidos a la actividad transactivadora. La unión a los elementos de respuesta glucocorticoide del ADN requiere la unión del RG activado en forma de homodímero, mientras que la interacción con los factores de transcripción como el AP-1 y el NF-κB se produce con el RG en forma de monómero. Estos efectos disociados se han demostrado mediante la creación de mutaciones del RG en células que se infectan con un vector vírico. Varias moléculas glucocorticoides, como la RU24858, la RU486 y la ZK98299, tienen mayor efecto de transrepresión que de transactivación. De hecho, los glucocorticoides inhalados que hoy día se utilizan para el tratamiento del asma y la EPOC, como el propionato de fluticasona o la budesonida, tienen un mayor efecto transrepresor, lo que justifica su potencia antiinflamatoria.
Recientemente se ha descrito una nueva clase de glucocorticoides con una potente actividad transrepresora y una mínima actividad transactivadora. Estos esteroides disociados, como el RU2458, el RU40066 o el activador selectivo no esteroideo ZK216348, muestran una potente actividad antiinflamatoria, que puede llegar a ser comparable a la de la prednisona, con un perfil de efectos adversos mucho menor que los glucocorticoides habituales. Esto indica que el desarrollo de nuevas moléculas de glucocorticoides con mayor margen de seguridad es posible, y que ello puede conducir al descubrimiento de esteroides orales sin efectos adversos significativos.
Antagonistas de los mediadores inflamatorios
Como ya se ha señalado, muchos mediadores (incluidos mediadores lipídicos y citocinas) intervienen en la patogenia de la EPOC. Varios antagonistas de estos mediadores se encuentran en fase de estudio clínico. Los anticuerpos contra el TNF-α (Infliximab, Remicade) están utilizándose para tratar a pacientes con artritis reumatoide y enfermedad inflamatoria intestinal, pero los efectos demostrados en la EPOC han sido hasta ahora decepcionantes.
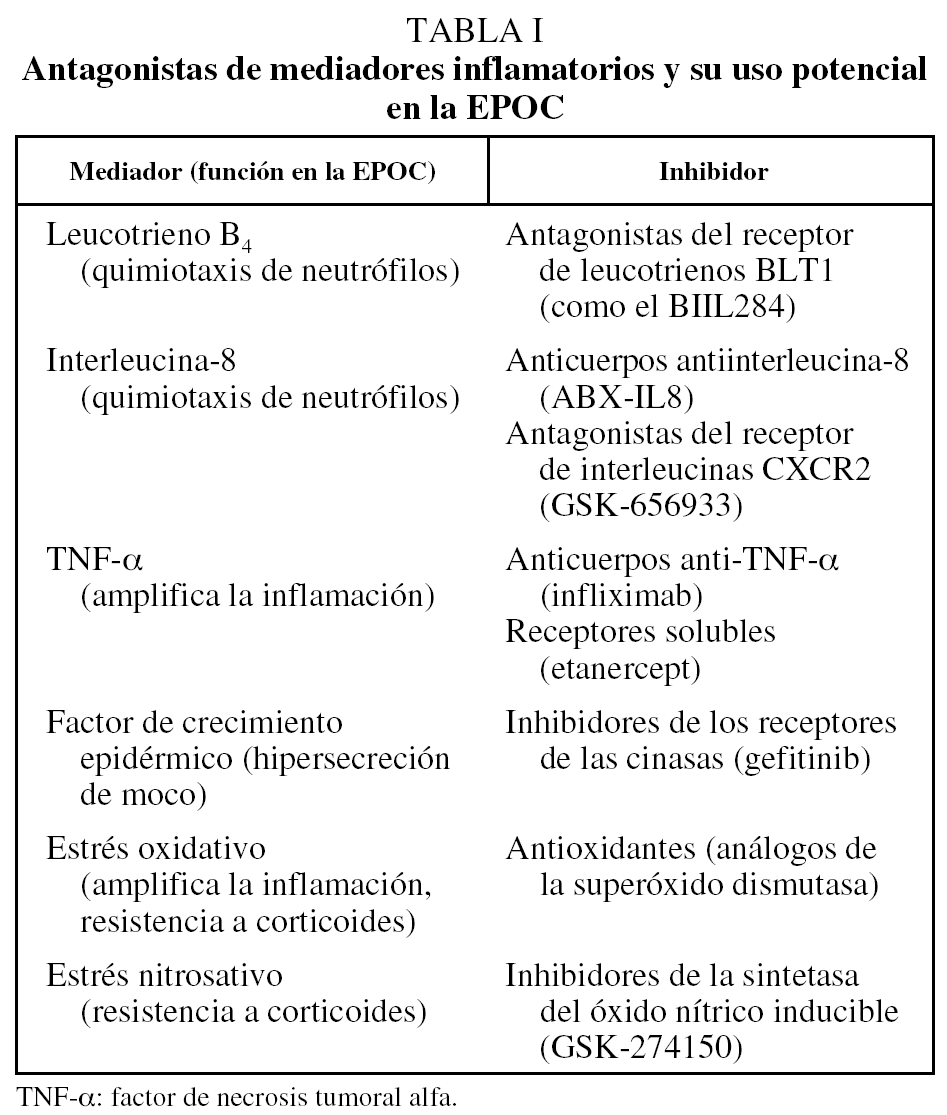
Otros antagonistas de mediadores inflamatorios se muestran en la tabla I.

Nuevos tratamientos antiinflamatorios
Entre los nuevos fármacos antiinflamatorios más prometedores en la EPOC (tabla II) se encuentran los inhibidores de la fosfodiesterasa-4 (PDE-4). Estos fármacos son activos por vía oral, incrementan las concentraciones de adenosinmonofosfato cíclico en las células inflamatorias y tienen efecto antiinflamatorio de amplio espectro tanto sobre células estructurales (fibroblastos, músculo liso, epitelio, glándulas) como sobre las células inflamatorias (macrófagos, neutrófilos, células T, eosinófilos). Sin embargo, aunque efectivos sobre modelos animales, no habían demostrado una gran eficacia en ensayos clínicos debido a la limitación de la dosis por los efectos adversos, sobre todo gastrointestinales, que presentaban. En un reciente estudio aleatorizado y controlado en pacientes con EPOC y volumen espiratorio forzado en el primer segundo del 50%, el roflumilast demostró una reducción significativa de las exacerbaciones y mejoría de la función pulmonar a las 24 semanas31, aunque alrededor del 5% de los pacientes presentaron diarrea. Se están investigando inhibidores de la PDE-4 más selectivos (inhibidores de la PDE-4B) y por vía inhalada para limitar sus efectos adversos.

Equilibrio proteasas-antiproteasas
El enfisema surge de la degradación de la matriz extracelular del pulmón. Esta matriz está compuesta por fibras elásticas formadas principalmente por colágeno tipo IV, proteoglucanos y elastina. La degradación principalmente de la elastina es la que da lugar a la formación del enfisema. Durante mucho tiempo se han propuesto varias proteasas que serían responsables de la degradación del tejido conectivo para el desarrollo del enfisema pulmonar. La elastasa y la proteasa-3, serinproteasas procedentes de los neutrófilos y las catepsi-nas han sido objeto de estudio durante muchos años y se han demostrado capaces de producir enfisema en modelos animales. La elastasa de los neutrófilos es inhibida por la alfa-1-antitripsina, lo cual explica el desarrollo de enfisema en los pacientes con deficiencia de esta enzima. Sin embargo, el papel en el enfisema asociado al humo del tabaco es menos claro. No obstante, existe cada vez más evidencia del papel de las metaloproteinasas (MMP) en la etiopatogenia del enfisema pulmonar32. Las MMP son una familia de más de 20 enzimas degradantes de la matriz extracelular derivadas de macrófagos y neutrófilos, y se piensa que son importantes para el desarrollo normal y la reparación del pulmón. La expresión anormal de estas enzimas se ha encontrado en otros procesos destructivos como la invasión tumoral y la angiogenia, artritis, arteriosclerosis o aneurismas arteriales, además de en el enfisema pulmonar. Los pacientes con enfisema presentan una mayor concentración en el lavado broncoalveolar y una mayor expresión en macrófagos alveolares de MMP-1 (colagenasa) y MMP-9 (gelatinasa B). También se ha observado un incremento de la actividad de la MMP-9 y la MMP-2 en el parénquima pulmonar de pacientes con enfisema. El interés por las MMP se ha visto incentivado tras la demostración de que la deleción en el gen que codifica la MMP-12 (macrófago metaloelastasa) previene el desarrollo de enfisema tras exposición al humo del tabaco en el ratón transgénico. Churg et al33 han demostrado recientemente que la inflamación producida por el humo del tabaco está mediada por la MMP-12, que actúa induciendo la liberación de TNF-α de los macrófagos, con el consiguiente efecto de activación endotelial, atracción de neutrófilos al pulmón y digestión de la matriz intersticial por las proteasas derivadas de los neutrófilos. Estos autores señalan que la liberación de TNF-α puede ser el mecanismo general por el cual el humo del tabaco produce inflamación pulmonar.
Normalmente todas estas enzimas elastolíticas están contrarrestadas por antiproteasas. La alfa-1-antitripsina en el parénquima y el SLPI en las vías aéreas son importantes inhibidores de las serinproteasas. Las metaloproteinasas tienen tres enzimas que inhiben su efecto, llamadas inhibidores tisulares de las metaloproteinasas (TIMP-1, TIMP-2 y TIMP-3). El humo del tabaco puede inducir inflamación y producir un aumento de la liberación de proteasas, que en sujetos sanos, en condiciones normales, quedan contrarrestadas por un incremento de antiproteasas en cantidad suficiente para evitar el daño en el pulmón. Sin embargo, en los fumadores que desarrollan EPOC la producción de antiproteasas puede ser inadecuada para neutralizar los efectos de las múltiples proteasas, probablemente debido a polimorfismos genéticos que alteran la función o la cantidad de estas enzimas.
Tratamiento con inhibidores de las proteasas
Las proteasas que intervienen en la destrucción alveolar podrían ser dianas terapéuticas para tratar a los pacientes con enfisema. Sin embargo, su inhibición por antiproteasas, bien endógenas como la alfa-1-antitripsina u otras pequeñas moléculas inhibidoras, no ha mostrado ninguna eficacia en estudios clínicos.
Estrés oxidativo
El humo del tabaco es una fuente de radicales libres, calculándose una cantidad de 1017 moléculas oxidantes por cada inhalación de un cigarrillo. Cada vez hay más evidencia de la implicación del estrés oxidativo en la patogenia de la EPOC34, aunque en el momento presente no existe un indicador directo de éste y los datos publicados corresponden a mediciones indirectas. Se ha demostrado un aumento de peróxido de hidrógeno en el condensado respiratorio de pacientes con EPOC, especialmente durante las exacerbaciones de la enfermedad35, y también un aumento de 8-isoprostanos, un marcador de oxidación lipídica, en condensado respiratorio y orina de pacientes con EPOC. Sin embargo, este exceso de carga oxidante no sólo proviene del tabaco, sino que además los macrófagos alveolares y los neutrófilos de los fumadores liberan más radicales de oxígeno que los de los no fumadores36. Por otro lado, las frecuentes exacerbaciones que experimentan estos pacientes, en gran parte secundarias a infecciones, pueden contribuir al reclutamiento y la activación de células fagocíticas al pulmón y aumentar la carga oxidante.
Los mecanismos por los que los oxidantes pueden lesionar el pulmón son variados. Por un lado, pueden alterar la actividad enzimática antielastolítica de los mecanismos contrarreguladores de la degradación del pulmón, como la alfa-1-antitripsina, cuya actividad está disminuida en fumadores. Otras antiproteasas, como el SLPI, también están reducidas en pulmones de fumadores y pacientes con EPOC. Los oxidantes podrían ser asimismo responsables directos de daño en las vías aéreas mediante la hipersecreción mucosa, edema alveolar y broncoconstricción. Además, el humo del tabaco puede inducir la activación del NF-κB y así activar la transcripción de mediadores de la inflamación, como el TNF-α y la IL-81. Estudios recientes han demostrado en fumadores y pacientes con EPOC una mayor expresión en el epitelio bronquial de la fracción p65 del NF-κB, que se correlaciona con la gravedad de la obstrucción al flujo aéreo37.
Las nuevas técnicas de biología molecular han facilitado la identificación de nuevos mecanismos de acción inflamatoria del estrés oxidativo, mediante la inactivación de enzimas con función antiinflamatoria, como las HDAC, que podría ser responsables de una respuesta inflamatoria amplificada y una disminución de la sensibilidad a glucocorticoides20.
Tratamiento antioxidante
El estrés oxidativo contribuye a amplificar la inflamación y puede ser responsable de la resistencia a los efectos antiinflamatorios de los corticoides, por lo que es una diana importante para futuros tratamientos. Los antioxidantes actualmente disponibles no han demostrado ser eficaces, pero nuevos antioxidantes más potentes y estables (como los análogos de la superóxido dismutasa) están en investigación.
Apoptosis, reparación y remodelación
Se dispone de evidencia científica cada vez más convincente de que la apoptosis desempeña un papel importante en la patogenia de la EPOC. Estudios en animales y en humanos han demostrado un incremento de la apoptosis en células estructurales (endoteliales, epiteliales y fibroblastos) de pacientes con EPOC en comparación con fumadores y sanos38.
La apoptosis es un mecanismo sumamente regulado que lleva a la muerte programada de las células. Es fundamental para el mantenimiento de la homeostasis de los tejidos y se halla en equilibrio con la proliferación y diferenciación. Hay tres vías moleculares implicadas en la apoptosis. Una de ellas se activa en respuesta a señales extracelulares y está mediada por miembros de la familia del TNF-α (Fas, TNF), unidos a receptores de superficie (death receptors), que dan lugar a activación de caspasas y liberación de desoxirribonucleasa. La segunda vía es intrínseca en respuesta a estrés químico o mecánico y da lugar a la liberación de citocromo C por la mitocondria, que a su vez activa las caspasas. La otra vía se activa en respuesta a estímulos citolíticos externos, como la formación de poros en la membrana por las perforinas y granzimas B liberadas por los linfocitos T.
Varios trabajos han descrito un aumento de células apoptóticas en tejido pulmonar enfisematoso junto a una mayor proliferación celular, lo que iría a favor de un intercambio o turnover celular alterado. En los casos en que se describe apoptosis de células endoteliales, ésta se ha asociado a una expresión reducida de factores de crecimiento del endotelio vascular, lo que ha dado lugar a la hipótesis vascular del enfisema, por la que la reducción de los factores de mantenimiento vascular produce la muerte de las células del endotelio alveolar y genera la destrucción alveolar que se observa en el enfisema39. Recientemente se ha postulado la existencia de un programa de mantenimiento del pulmón con un equilibrio entre la capacidad reparadora en respuesta al daño ambiental y la destrucción alveolar por apoptosis y estrés oxidativo. La edad puede ser igualmente determinante en el fracaso de este programa de mantenimiento, lo que puede convertirla en un factor de riesgo independiente para el desarrollo de enfisema. De nuevo, el factor de crecimiento del endotelio vascular (VEGF) se ha propuesto como un factor de mantenimiento de la homeostasis de la matriz extracelular y la arquitectura alveolar. La interacción de la apoptosis con los otros mecanismos patogénicos de la EPOC se expresa en la figura 1.
Otro tipo de respuesta celular al estrés es la senescencia replicativa, fenómeno que aparece con la edad y que puede acelerarse por el estrés oxidativo, lo que podría alterar la reparación de tejido bloqueando la regeneración celular, disminuyendo las células progenitoras y alterando la estructura del órgano mediante la destrucción de la matriz extracelular. En pacientes con enfisema pulmonar se han demostrado mayores índices de senescencia en las células epiteliales y endoteliales alveolares40, lo que podría explicar una reparación deficiente.
Tratamiento regenerador del tejido pulmonar: células madre
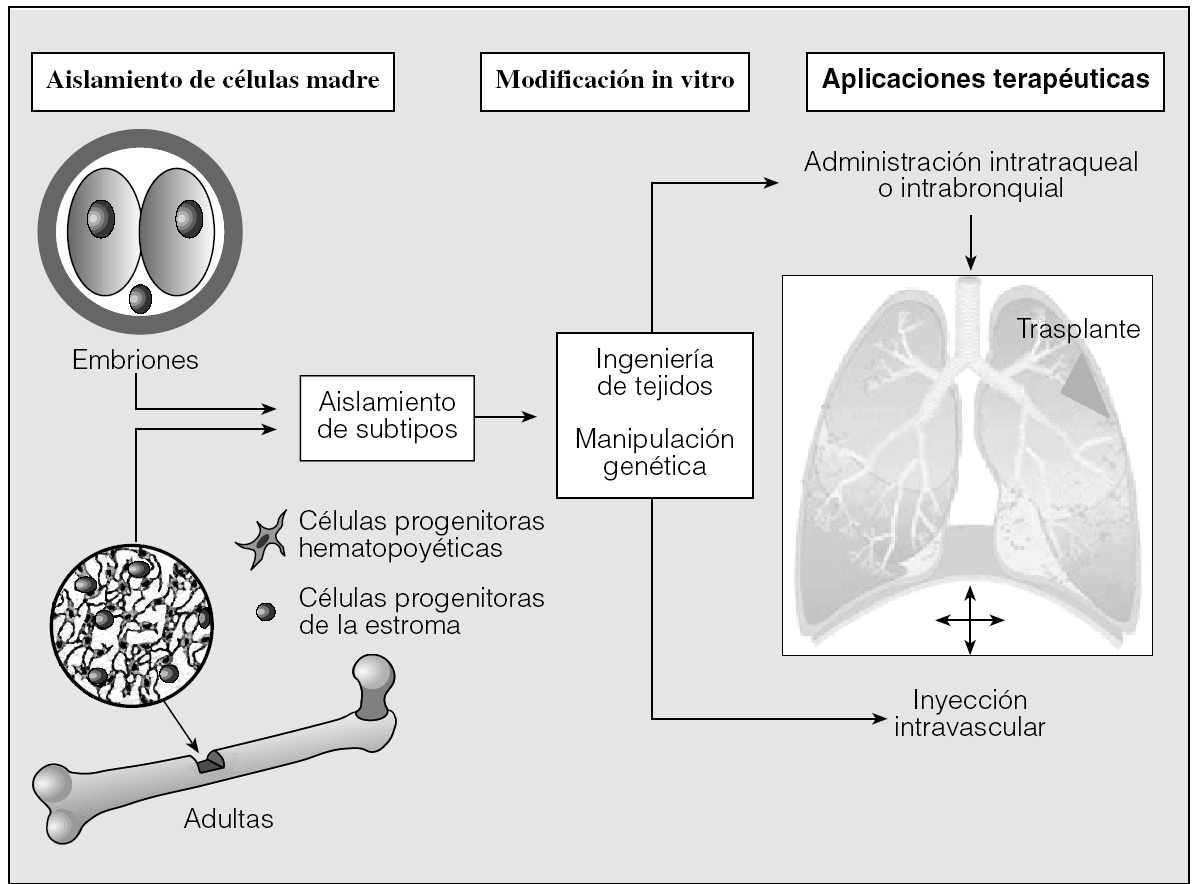
Con el creciente conocimiento de la biología de las células progenitoras, su potencial capacidad regeneradora del pulmón está adquiriendo cada vez mayor interés. En modelos animales hay evidencia de que las células progenitoras de la médula ósea pueden anidar en el pulmón y repararlo. Este hecho abre la posibilidad de expresar genes en células progenitoras manipuladas para evitar una reacción inmunológica. Otra posible aplicación consistiría en diferenciar estas células progenitoras in vitro antes de trasplantarlas al pulmón (ingeniería de tejidos), aunque este proceso sólo podría realizarse clonando células embriógenas, lo que de momento plantea conflictos éticos (fig. 2). Las células madre adultas son más difíciles de aislar y cultivar, aunque se ha demostrado su multipotencialidad in vitro. Con independencia del tipo de células utilizadas, los criterios para un tratamiento eficaz deberían ser los siguientes: que las células adopten el fenotipo y la función adecuados; control preciso de la expresión de los genes manipulados; que las células aniden en el pulmón el tiempo necesario y suficiente para reparar el daño, y que carezcan de capacidad para malignizarse41.

Fig. 2. Aplicaciones futuras del tratamiento de reparación con células madre. (Modificada de Griffiths et al41.)
La diferenciación de las células progenitoras de la médula ósea en células epiteliales contribuye a la reparación del daño diario, pero en caso de lesiones más graves esta reparación puede incrementarse por células pluripotenciales autólogas genéticamente manipuladas o exógenas. Yamada et al42 estudiaron el papel de células progenitoras de la médula ósea marcadas en la reparación del pulmón en un modelo animal de enfisema y uno de lesión pulmonar con lipopolisacárido. En ambos modelos encontraron en las paredes alveolares células marcadas de la médula ósea que adoptaban fenotipos de células endoteliales y neumocitos tipo 1. Tras manipular la liberación de estas células por la médula, estimulándola con factores estimuladores de las colonias granulocíticas o suprimiéndola con radiación no letal de la médula, confirmaron la hipótesis de la contribución de la médula en la reparación pulmonar.
Conclusiones
La mejora del conocimiento de los mecanismos que subyacen al desarrollo de la EPOC está ayudando al desarrollo de nuevos tratamientos y abre un mundo de posibilidades para mejorar el cuidado y el pronóstico de esta enfermedad. Avanzar en la investigación básica de estos mecanismos y ligarla con las necesidades clínicas es nuestro reto para los años venideros.
Correspondencia: Dr. B.G. Cosío.
Servicio de Neumología. Hospital Universitario Son Dureta.
Andrea Doria, 55. 07014 Palma de Mallorca. Islas Baleares. España.
Correo electrónico: bcosio@hsd.es







